Este método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde. Al final del proceso se obtendrán 2 moléculas de ADN, cada una de ellas formada por una cadena original y una cadena complementaria que ha sido sintetizada. La enzima que realiza este proceso se denomina DNA polimerasa. Esta enzima añade los nucleótidos en el hidroxilo 3 terminal de la cadena de DNA paterna.
ETAPAS:
A)Desnaturalización: Se comienza calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrógeno y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación.
B)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN. 2- Enzima de restricción (BRENDA)
Es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
El mecanismo de corte de DNA se realiza a través de la ruptura de dos enlaces fosfodiéster en la doble hebra, lo que da lugar a dos extremos de DNA. Éstos pueden ser romos (cuando los enlaces rotos coinciden) o Cohesivos/escalonados. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
3 - ELISA… El ensayo inmunoabsorbente ligado a enzimas (ELISA) utiliza como sus siglas lo indican una enzima como marcador para mediar la formación de complejos antígeno-anticuerpo. Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario. Casi todas las pruebas ELISA son ensayos en fase sólida en los cuales se adsorbe un antígeno o un anticuerpo sobre un soporte sólido. Algunos de los protocolos se basan en reacciones de enlace competitivo y otras en reacciones de enlace no competitivo, pero en todas las pruebas ELISA se requiere de un paso de separación para eliminar el conjugado enzimático libre antes de proceder a determinar la cantidad de conjugado enzimático enlazado. Para lo cual se añade sustrato enzimático y se mide la reacción catalítica entre la enzima y el sustrato. Por sus características catalíticas las enzimas son marcadores muy sensibles y versátiles. Una sola proteína enzimática puede transformar en algunos minutos gran número de moléculas de sustrato en una cantidad igualmente abundante de producto final, produciendo un cambio de color amplificado y que se detecta con facilidad.En el método ELISA de inhibición el cambio de color se reduce, porque la actividad enzimática se inhibe cuando el anticuerpo se enlaza al conjugado enzimático.
Se considera una extencion de la tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma. 4 - Southern Blot para ADN…
Southern blot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda. Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern. El sector de ADN buscando puede ser un gen, o parte del mismo. Se puede utilizar para el diagnostico molecular de patologias geneticas. LA doble hebra de ADN se forma por complementariedad SONDA mustra (una de las hebras es la sonda en simple caden y la otra es la muestra en simple cadena). Los nucleotidos de la sonda esta macados P 32 (radioactivos); si al revelar hay marcador radioactivo presente, esta en la muestra analizada la secuencia complementaria a la sonda. Pasos: extraccion de ADN. Fragmentar el ADN en porciones mas pequeñas (enzimas de resctriccion). Separar los ragmentos realizando una corrida electroforetica usando como sustrato de migracion un gel. (separacion por tamaño). Se agrega la sonda sobre la membrana y se incuba. Se lava para quitar el maximo de hibridaciones inespecificas. Se revela por autoradiografia.
5- Northern Blot para RNA
Northern blot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente. El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southern blot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northern blot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.
El western blot es un método en biología molecular/bioquímica/inmunogenética para la detección de proteínas en una muestra de un tejido homogeneizado o extracto. Se utiliza una electroforesis para separar proteínas desnaturalizadas de la masa. Las proteínas son trasferidas desde el gel hacia la membrana (originalmente de nitrocelulosa), donde son examinadas utilizando anticuerpos específicos para la proteína. Como resultado, se pueden examinar la cantidad de proteínas en una muestra y comparar los niveles de presencia entre varios grupos. Otras técnicas ,también usando anticuerpos, permiten la detención de proteínas en tejidos (inmunohistoquímica) y células (inmunocitoquímica). En el Northern blot, se utiliza para identificar las cadenas de ARN de secuencia semejante al ADN que se usa como sonda; a diferencia del tipo Southern que se utiliza para identificar ADN, el método northern sirve para identificar ARN. De manera resumida, el Western sirve para identificar proteínas y el Northern para identificar secuencias de RNA. Bueno, sus aplicaciones son muy variadas y un ejemplo que puedo decirte es el de la detección del VIH. De manera simple, el western blot se utiliza para detectar las proteínas de la membrana del virus, como dice en la descripción de arriba, estas proteínas provienen de la muestra (generalmente sangre que ya está infectada con el virus), las cuales son separadas por electroforesis en gel de poliacrilamida, y posteriormente detectadas por un anticuerpo específico (específico a la proteína, es decir, que sólo podrá unirse a esa proteína, a ninguna otra) en la membrana de nitrocelulosa. Bueno, espero que te sirva de algo, lo primero lo saqué de wikipedia, pero me pareció muy escueto, por lo que decidí ampliarlo un poquito con lo que me acuerdo. Ah, si, y el northern detecta secuencias de RNA, y tiene aplicaciones similares, ya que como sabes, cuando hay síntesis de proteínas, existe un RNA mensajero que, junto con el RNA ribosomal y el de transferencia, que sintetizan a la proteína en el retículo endoplásmico... entonces también pueden detectarse un RNA mensajero específico para una proteína de interés, como en el caso del VIH...
8-Southern Blot para adn y proteina El Southern Blot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizando sonicación ó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAds ó RNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.El traspaso de las muestras desde el gel al filtro es una etapa esencial, porque el reconocimiento debases complementarias entre sonda y secuencias de DNA en el gel es muy ineficiente.Cabe señalar que el nombre de la técnica Southern Blot deriva en parte del apellido Southern delinvestigador que la desarrolló y de la palabra en inglés Blot que significa traspasar
El ADN mitocondrial (mtDNA) es un material genético circular cerrado de doble cadena que se localiza en el interior de las mitocondrias celulares.El genoma mitocondrial consta de aproximadamente 16500 pares de bases (p.b.), y codifica para una pequeña fracción de las proteínas mitocondriales. El mtDNA contiene información de 38 genes: 2rRNA (12S y 16S), 22tRNA y 13 genes estructurales, los cuales codifican diferentes subunidades de los complejos enzimáticos del sistema de fosforilación oxidativa.La región mayor no codificante, conocida como región control o D-Loop, ocupa 1122 pares de bases. Esta región destaca por su elevada tasa de mutación y por ser muy variable entre las diferentes poblaciones. La variabilidad en la región control se concentra básicamente en tres regiones o segmentos hipervariables:
- HVSI (posiciones 16024-16365)
- HVSII (posiciones 73-340)
- HVSIII (posiciones 438-574)
De las tres, la más polimórfica es la HVSI, por lo que es la que se analiza principalmente en estudios de Antropología, Genética de Poblaciones y Medicina Forense.
El estudio del ADN mitocondrial resulta especialmente recomendado cuando se trabaja con muestras muy degradadas, como es de esperar en ADN antiguo. Se calcula que una célula puede contener hasta un centenar de mitocondrias, y que dentro de cada mitocondria coexisten entre 1000 y 10000 copias de ADN mitocondrial. Este elevado número de moléculas de ADN mitocondrial en la célula hace que su recuperación en aquellos casos en los que el ADN de partida es muy escaso o está muy degradado, sea mucho más eficiente que, por ejemplo, la de ADN nuclear o autosómico. En la especie humana, el sexo viene determinado por el tipo de cromosomas sexuales presentes en las células (cromosomas X e Y). Así, el sexo femenino viene definido por dos cromosomas X (XX) y el masculino por un cromosoma X y un cromosoma Y (XY). A excepción de dos pequeñas regiones situadas en los extremos en las que existe cierto grado de recombinación con el cromosoma X, el cromosoma Y se transmite como una unidad íntegra de padres a hijos varones. Esta característica hace que, al igual que el ADN mitocondrial para las mujeres, pueda seguirse una determinada variante o haplotipo en sucesivas generaciones. El Cromosoma Y se analiza de forma rutinaria en el campo de la Genética de Poblaciones y la Genética Forense. Sin embargo, su empleo en estudios de ADN antiguo es todavía muy restringido por motivos técnicos que tienen que ver con su escasez, dado que únicamente existe una copia de cromosoma Y por célula.
10- Electroresis de proteina Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína.
La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL. A usted le pueden solicitar no comer ni beber nada durante 12 horas antes de un examen de electroforesis de lipoproteínas.
El médico le puede solicitar que deje de tomar medicamentos que pudieran afectar el examen. NO suspenda ningún medicamento sin consultarlo previamente con éste.
Los medicamentos que pueden afectar la medición de las proteínas totales incluyen clorpromazina, corticoesteroides, isoniazida, neomicina, fenacemida, salicilatos, sulfamidas y tolbutamida.Cuando se inserta la aguja para extraer la sangre, algunas personas sienten un dolor moderado, mientras que otras sólo sienten un pinchazo o sensación de picadura. Posteriormente, puede haber algo de sensación pulsátil.Las proteínas están compuestas de aminoácidos y son componentes importantes de todas las células y los tejidos. Existen muchas clases de proteínas en el cuerpo, con muchas funciones diferentes. Los ejemplos de proteínas son: las enzimas, algunas hormonas, la hemoglobina, la lipoproteína de baja densidad (colesterol "malo") y otras.
Las proteínas séricas se clasifican como albúmina y globulinas. La albúmina es la proteína de mayor concentración en el suero. Trasporta muchas moléculas pequeñas, pero también es importante para impedir que el líquido se escape de los vasos sanguíneos hacia los tejidos.
La Reacción en Cadena de la Polimerasa (PCR) se realiza en el laboratorio con el objetivo de obtener un gran número de copias de un fragmento de ADN a partir de una pequeña muestra de la original. También nos permite amplificar la cadena, de manera que sea más fácil identificar algún virus o enfermedad dentro de estas. Costa de tres fases: 1) Desnaturalización (se calienta la muestra para romper las cadenas), 2) Templado (bajar la temperatura para que los iniciadores se unan con otras cadenas) y 3) Polimerización (se eleva de nuevo la temperatura para copiar el acido nucleído). Una enzima de restricción es laque puedes reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortarlo (atreves de la ruptura de dos enlaces fosfodiester en la doble hebra) en el sitio de restricción o no muy lejano a este. Estos sitios cuentan con 4 a 12 pares de bases. Los fragmentos que resultan del corte se pueden unir con otras enzimas llamada ligasas. La Enzima de Restricción o endonucleasa de restricción) se conoce como BRENDA en sus siglas en ingles Bacterial Restriction Endonuclease Analysis. ELISA (del ingles Enzyme-Linked ImmunoSorbent Assay, Ensayo por inmunoabsorción ligado a enzimas) es una técnica de inmuno-ensayo en la cual un antígeno inmovilizado se detecta mediante un anticuerpo enlazado a una enzima capaz de generar un producto detectable como cambio de color o algún otro tipo. La aparición de colorantes permite medir indirectamente mediante espectrofotometría el antígeno en la muestra. Es el examen utilizado para la detección del VIH en el ADN. La Técnica de Sorthern se emplea para identificar secuencias específicas de DNA, mediante rompimiento con enzimas de restricción. Los fragmentos obtenidos se someten a electroforesis en un gel de agarosa, donde los fragmentos de mayor tamaño quedaran en la parte superior y las de menor tamaño en la parte inferior. La técnica de Northen se emplea para identificar secuencias específicas de RNA. Consiste en aislarlo del citoplasma o del núcleo y se somete a electroforesis en gel de agarosa en condiciones desnaturalizantes para que los RNA se separen según su clase o peso molecular. La electroforesis, mencionada en casi todas las técnicas anteriores, nos ayuda a observar los ácidos nucleicos y proteínas al final del procedimiento. En él las moléculas migran a través de un gel en el cual, por acción de un campo eléctrico, serán separadas de acuerdo a su peso molecular.
)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN. 2- Enzima de restricción (BRENDA)
Es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
Tema 1: REACCIÓN EN CADENA DE LA POLIMERASA (PCR) Este método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde.
ETAPAS: A) Desnaturalización: Se comienza calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrógeno y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos.
B)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.
Rocio Garcia .............2012-1235 Reaccion de cadena polimerasa
La Reacción en Cadena de la Polimerasa (PCR) es una técnica "in vitro" que imita la habilidad natural de la célula de duplicar el ADN. Se trata de una técnica usada para crear un gran número de copias de un segmento de ADN, que utiliza ciclos de desnaturalización, apareamiento con cebadores y extensión por una ADN polimerasa termoresistente. El único método para obtener grandes cantidades de un fragmento de ADN era clonándolo en vectores adecuados e introduciéndolo y multiplicándolo en bacterias. En el año 1985, un investigador norteamericano, Kary Mullis (acreedor del Premio Nobel en Química 1993 por este aporte), desarrolló un método que permite, a partir de una muestra muy pequeña de ADN, obtener millones de copias de ADN in vitro, en unas pocas horas y sin necesidad de usar células vivas. Esta técnica, llamada reacción en cadena de la polimerasa (PCR), requiere conocer la secuencia de nucleótidos de los extremos del fragmento que se quiere amplificar. Estas secuencias se usan para diseñar dos oligonucleótidos sintéticos de ADN complementarios a una porción de cada una de las dos cadena de la doble hélice.
Tema 1: REACCIÓN EN CADENA DE LA POLIMERASA (PCR) Este método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde. Al final del proceso se obtendrán 2 moléculas de ADN, cada una de ellas formada por una cadena original y una cadena complementaria que ha sido sintetizada. La enzima que realiza este proceso se denomina DNA polimerasa. Esta enzima añade los nucleótidos en el hidroxilo 3 terminal de la cadena de DNA paterna. ETAPAS: A) Desnaturalización: Se comienza calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrógeno y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación. B)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma. C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
tema 2- enzima de restricción (brenda) es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de adn y cortar el adn en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos. el mecanismo de corte de dna se realiza a través de la ruptura de dos enlaces fosfodiéster en la doble hebra, lo que da lugar a dos extremos de dna. éstos pueden ser romos (cuando los enlaces rotos coinciden) o cohesivos/escalonados. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
Tema 3 - ELISA… El ensayo inmunoabsorbente ligado a enzimas (ELISA) utiliza como sus siglas lo indican una enzima como marcador para mediar la formación de complejos antígeno-anticuerpo. Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario. Casi todas las pruebas ELISA son ensayos en fase sólida en los cuales se adsorbe un antígeno o un anticuerpo sobre un soporte sólido. Algunos de los protocolos se basan en reacciones de enlace competitivo y otras en reacciones de enlace no competitivo, pero en todas las pruebas ELISA se requiere de un paso de separación para eliminar el conjugado enzimático libre antes de proceder a determinar la cantidad de conjugado enzimático enlazado. Para lo cual se añade sustrato enzimático y se mide la reacción catalítica entre la enzima y el sustrato. Por sus características catalíticas las enzimas son marcadores muy sensibles y versátiles. Una sola proteína enzimática puede transformar en algunos minutos gran número de moléculas de sustrato en una cantidad igualmente abundante de producto final, produciendo un cambio de color amplificado y que se detecta con facilidad.En el método ELISA de inhibición el cambio de color se reduce, porque la actividad enzimática se inhibe cuando el anticuerpo se enlaza al conjugado enzimático. Tema4 - Southern Blot para ADN… Southernblot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda. Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern. El sector de ADN buscando puede ser un gen, o parte del mismo. Se puede utilizar para el diagnostico molecular de patologiasgeneticas. LA doble hebra de ADN se forma por complementariedad SONDA mustra (una de las hebras es la sonda en simple caden y la otra es la muestra en simple cadena). Los nucleotidos de la sonda esta macados P 32 (radioactivos); si al revelar hay marcador radioactivo presente, esta en la muestra analizada la secuencia complementaria a la sonda. Pasos: extraccion de ADN. Fragmentar el ADN en porciones mas pequeñas (enzimas de resctriccion). Separar los ragmentos realizando una corrida electroforetica usando como sustrato de migracion un gel. (separacion por tamaño). Se agrega la sonda sobre la membrana y se incuba. Se lava para quitar el maximo de hibridaciones inespecificas. Se revela por autoradiografia.
Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN. 2- Enzima de restricción (BRENDA)
Es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
Tema 5-NORTHERNBLOT PARA RNA Northernblot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente. El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southernblot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northernblot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.
TEMA 6-EASTERN BLOT Se considera una extencion de la tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma. pueden examinar la cantidad de proteínas en una muestra y comparar los niveles de presencia entre varios grupos. Otras técnicas ,también usando anticuerpos, permiten la detención de proteínas en tejidos (inmunohistoquímica) y células (inmunocitoquímica). En el Northernblot, se utiliza para identificar las cadenas de ARN de secuencia semejante al ADN que se usa como sonda; a diferencia del tipo Southern que se utiliza para identificar ADN, el método northern sirve para identificar ARN.
TEMA 7-WESTERN BLOT PARA PROTEINA EL WESTERN BLOT ES UN MÉTODO EN BIOLOGÍA MOLECULAR/BIOQUÍMICA/INMUNOGENÉTICA PARA LA DETECCIÓN DE PROTEÍNAS EN UNA MUESTRA DE UN TEJIDO HOMOGENEIZADO O EXTRACTO. SE UTILIZA UNA ELECTROFORESIS PARA SEPARAR PROTEÍNAS DESNATURALIZADAS DE LA MASA. LAS PROTEÍNAS SON TRASFERIDAS DESDE EL GEL HACIA LA MEMBRANA (ORIGINALMENTE DE NITROCELULOSA), DONDE SON EXAMINADAS UTILIZANDO ANTICUERPOS ESPECÍFICOS PARA LA PROTEÍNA. COMO RESULTADO, SE PUEDEN EXAMINAR LA CANTIDAD DE PROTEÍNAS EN UNA MUESTRA Y COMPARAR LOS NIVELES DE PRESENCIA ENTRE VARIOS GRUPOS. OTRAS TÉCNICAS ,TAMBIÉN USANDO ANTICUERPOS, PERMITEN LA DETENCIÓN DE PROTEÍNAS EN TEJIDOS (INMUNOHISTOQUÍMICA) Y CÉLULAS (INMUNOCITOQUÍMICA). EN EL NORTHERNBLOT, SE UTILIZA PARA IDENTIFICAR LAS CADENAS DE ARN DE SECUENCIA SEMEJANTE AL ADN QUE SE USA COMO SONDA; A DIFERENCIA DEL TIPO SOUTHERN QUE SE UTILIZA PARA IDENTIFICAR ADN, EL MÉTODO NORTHERN SIRVE PARA IDENTIFICAR ARN. DE MANERA RESUMIDA, EL WESTERN SIRVE PARA IDENTIFICAR PROTEÍNAS Y EL NORTHERN PARA IDENTIFICAR SECUENCIAS DE RNA.
TEMA8-SOUTHERN BLOT PARAADN Y PROTEINA El SouthernBlot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizandosonicaciónó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAdsóRNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.
Tema 9- Extracción de adn mitocondrial El ADN mitocondrial (mtDNA) es un material genético circular cerrado de doble cadena que se localiza en el interior de las mitocondrias celulares.El genoma mitocondrial consta de aproximadamente 16500 pares de bases (p.b.), y codifica para una pequeña fracción de las proteínas mitocondriales. El mtDNA contiene información de 38 genes: 2rRNA (12S y 16S), 22tRNA y 13 genes estructurales, los cuales codifican diferentes subunidades de los complejos enzimáticos del sistema de fosforilaciónoxidativa.La región mayor no codificante, conocida como región control o D-Loop, ocupa 1122 pares de bases.
8-Southern Blot para adn y proteina El Southern Blot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizando sonicación ó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAds ó RNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.El traspaso de las muestras desde el gel al filtro es una etapa esencial, porque el reconocimiento debases complementarias entre sonda y secuencias de DNA en el gel es muy ineficiente.Cabe señalar que el nombre de la técnica Southern Blot deriva en parte del apellido Southern delinvestigador que la desarrolló y de la palabra en inglés Blot que significa traspasar
TEMA 10- ELECTRORESIS DE PROTEINA Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas. Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína. La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL.
La pureza de los reactivos es fundamental para la exactitud que se obtiene en cualquier análisis. En el laboratorio se dispone de distintos tipos de reactivos (sólidos, líquidos o disoluciones preparadas) tal y como se comercializan. En general, las casas comerciales ofrecen un mismo producto con varias calidades. Es importante que cuando seleccionemos un reactivo su calidad esté en concordancia con el uso que se le va a dar.
2.1. Clasificación
En el laboratorio de análisis se utilizan reactivos de calidad analítica que se producen comercialmente con un alto grado de pureza. En las etiquetas de los frascos se relacionan los límites máximos de impurezas permitidas por las especificaciones para la calidad del reactivo o los resultados del análisis para las distintas impurezas. Dentro de los reactivos analíticos pueden distinguirse tres calidades distintas:
• Reactivos para análisis (PA): Son aquellos cuyo contenido en impurezas no rebasa el número mínimo de sustancias determinables por el método que se utilice. • Reactivos purísimos: Son reactivos con un mayor grado de pureza que los reactivos “para análisis” . • Reactivos especiales: Son reactivos con calidades específicas para algunas técnicas analíticas, como cromatografía líquida (HPLC), espectrofotometría (UV)… Hay reactivos que tienen características y usos específicos como los reactivos calidad patrón primario, que se emplean en las técnicas volumétricas, o los patrones de referencia.
La reacción en cadena de la polimerasa, conocida como PCR por sus siglas en inglés (polymerase chain reaction), es una técnica de biología molecular desarrollada en 1987 porKary Mullis,1 cuyo objetivo es obtener un gran número de copias de un fragmento de ADNparticular, partiendo de un mínimo; en teoría basta partir de una única copia de ese fragmento original, o molde. Esta técnica sirve para amplificar un fragmento de ADN; su utilidad es que tras la amplificación resulta mucho más fácil identificar con una muy alta probabilidad, virus obacterias causantes de una enfermedad, identificar personas (cadáveres) o hacer investigación científica sobre el ADN amplificado. Estos usos derivados de la amplificación han hecho que se convierta en una técnica muy extendida, con el consiguiente abaratamiento del equipo necesario para llevarla a cabo.
ENZIMA DE RESTRICCIÓN Un enzima de restricción (o endonucleasas de restricción) es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos. El mecanismo de corte de DNA se realiza a través de la ruptura de dos enlaces fosfodiéster en la doble hebra, lo que da lugar a dos extremos de DNA. Éstos pueden ser romos (cuando los enlaces rotos coinciden) o Cohesivos/escalonados. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick). Los fragmentos de ADN obtenidos de este modo pueden unirse por otras enzimas llamadas ligasas. Las enzimas de restricción que a pesar de ser distintas y provenir de distintas especies, tienen la misma secuencia de reconocimiento y dejan el mismo extremo cohesivo, pero no cortan en el mismo sitio, son llamadas isoesquizómeros. Por ejemplo, están los isoesquizómeros Asp718 y KpnI. El Premio Nobel de Medicina de 1978 fue concedido a los microbiólogos Werner Arber, Daniel Nathans y Hamilton Smith por el descubrimiento de las endonucleasas de restricción lo que condujo al desarrollo de la tecnología de ADN recombinante. El primer uso práctico de su trabajo fue la manipulación de la bacteria E. coli para producir insulina humana para los diabéticos. Uno de los campos en los que las enzimas de restricción han tenido mayor implicación ha sido el diagnóstico de enfermedades genéticas relacionadas con cambios en la secuencia del ADN, ya sean mutaciones puntuales, inserciones o deleciones de fragmentos. Si éstas se producen en un sitio de reconocimiento de la enzima de restricción, al producirse eliminarán o agregarán nuevos sitios de corte. Al aplicar esta enzima al gen de una persona sana y una enferma se deberían observar distintas cantidades de fragmentos para cada caso en una electroforesis.
Elisa Directo Samdwis ELISA (acrónimo del inglés Enzyme-Linked ImmunoSorbent Assay, Ensayo por inmunoabsorción ligado a enzimas) es una técnica de inmunoensayo en la cual un antígeno inmovilizado se detecta mediante un anticuerpo enlazado a una enzima capaz de generar un producto detectable como cambio de color o algún otro tipo; en ocasiones, con el fin de reducir los costos del ensayo, nos encontramos con que existe un anticuerpo primario que reconoce al antígeno y que a su vez es reconocido por un anticuerpo secundario que lleva enlazado la enzima anteriormente mencionada. La aparición de colorantes permite medir indirectamente mediante espectrofotometría el antígeno en la muestra. Se usa en muchos laboratorios para determinar si un anticuerpo particular está presente en la muestra de sangre de un paciente. Aunque el procedimiento es rutinario y sencillo, involucra a un gran número de variables, tales como seleción de reactivo, temperatura, medición de volumen y tiempo, que si no se ajustan correctamente puede afectar a los pasos sucesivos y al resultado de la prueba. La interacción antígeno-anticuerpo en el laboratorio puede ser utilizada para determinar si un paciente tiene una infección o una enfermedad autoinmune. Pero como prueba diagnóstica, posee diversas limitaciones que conviene conocer: -En primer lugar, un resultado positivo que confirma la presencia de anticuerpos no significa necesariamente que el paciente esté enfermo. El cuerpo de una persona que ha estado enferma y que ya se ha recuperado, puede seguir produciendo anticuerpos. Esto originaría un falso positivo. -En segundo lugar, hay personas que producen una baja cantidad de anticuerpos, por lo que éstos pueden pasar desapercibidos y no ser medidos, dando lugar a un falso negativo. Sería el caso, por ejemplo, de personas que padezcan una inmunodeficiencia, o que se encuentren en el periodo ventana de la infección en el momento de realizar la prueba, o que estén infectadas por una cepa extraña. -En tercer lugar, pueden aparecer falsos positivos cuando se da inespecificidad entre antígeno-anticuerpo. En estos casos de diagnóstico de enfermedad es recomendable la eliminación (mediante centrifugación) de células de la sangre que puedan interferir con el ensayo y pueda ocasionar un resultado falso positivo, careciendo aquél de especificidad. También, debemos tener en cuenta que cuando se trata de diagnosticar una enfermedad que posee un valor predictivo positivo bajo en una determinada población, es decir, que esa enfermedad tiene muy baja incidencia en dicha población, es necesario volver a confirmar el resultado positivo mediante otro método de diagnóstico independiente. Normalmente, se lleva a cabo un western blot donde se detecta la presencia de varios anticuerpos, simultáneamente, frente a la misma infección en una muestra. El resultado del western se considera positivo cuando aparecen al menos 5 bandas, que indica que 5 anticuerpos diferentes están presentes en el sujeto frente a esa infección. Entonces es cuando se diagnostica como positivo a dicho paciente.
La técnica de ELISA es un ensayo inmunoenzimático ampliamente empleado en el área médica para la cuantificación de moléculas especialmente de aquellas que experimentan cambios en diferentes estados como pueden ser infecciones por bacterias, virus, hongos o parásitos o fases activas de enfermedades autoinmunes. Como ejemplo se pueden medir por ELISA hormonas, autoanticuerpos, inmunoglobulinas contra antígenos de patógenos, toxinas, antígenos. Las técnicas de ELISA son también muy usadas en los ensayos de nuevos medicamentos para comprobar sus efectos cuantificando las moléculas de interés en cada caso. Existen numerosas variantes de este tipo de ensayo. Entre ellos se encuentran los métodos directo e indirecto. El método directo permite la detección del antígeno con un anticuerpo específico conjugado con una enzima como sistema de marcaje. En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA.
TEMA 4 :Southern blot, Southern blot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda.1 Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern. 1. Extracción del ADN Se puede extraer ADN de casi cualquier tejido humano. Las posibles fuentes de ADN en la escena de un delito incluyen sangre, semen, tejido de una víctima muerta, células del folículo capilar y saliva. El ADN extraído de las pruebas del delito ("indicios" o "vestigios" biológicos) se compara con el extraído de muestras de referencia, obtenidas de personas conocidas,habitualmente de la sangre. 2. Digestión del ADN con una endonucleasa de restricción El ADN extraído de la muestra se trata con una endonucleasa de restricción,que es una enzima que corta el ADN bicatenario en donde tenga una secuencia característica. La enzima que se usa más frecuentemente para el análisis legal es HaeIII, que corta el ADN en la secuencia 5'-GGCC-3'. 3. Electroforesis en gel de agarosa Tras la digestión del ADN, los fragmentos de ADN resultantes se separan según su tamaño mediante electroforesis en geles de agarosa. Durante la electroforesis, las moléculas de ADN, que poseen carga negativa, migran hacia el electrodo positivo. Al avanzar las moléculas de ADN, su velocidad de migración se ve reducida por la matriz del gel de agarosa. Las moléculas menores se mueven más deprisa a través de los poros del gel que las de mayor tamaño. Como resultado, se produce una separación continua de los fragmentos de ADN de acuerdo con su tamaño, de modo que los fragmentos más pequeños avanzan la mayor distancia con referencia al origen o punto de aplicación de la muestra. 4. Preparación de un ensayo de Southern ("Southern blot") Tras la electroforesis, las moléculas de ADN separadas se desnaturalizan mientras permanecen en el gel de agarosa, impregnando éste con una disolución alcalina. Tras la neutralización de ésta, el DNA monocatenario resultante se transfiere a la superficie de una membrana de nailon,realizando así una copia o "calco" (la traducción más literal del inglés blot,conservando este sentido, es "secante"). Este proceso de desnaturalización y transferencia se conoce como método de Southern en recuerdo de quien lo inventó, Edward Southern. Al igual que la aplicación de un secante a un papel con la tinta húmeda transfiere una réplica de la imagen del papel al secante,el "calco" del ADN en el gel a la membrana de nailon conserva la distribución espacial de los fragmentos de ADN conseguida en el gel como resultado de la electroforesis.
TEMA 5 :NORTHERN BLOT Northern blot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente.1 El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southern blot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northern blot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford. PROCEDIMIENTO El procedimiento general comienza con la extracción del RNA total de una muestra de tejido homogenizado. El mRNA se puede aislar a través del uso de cromatografía para mantener solamente los ARN con colas de poli-A. Las muestras de ARN son entonces separadas por electroforesis en gel. Dada la fragilidad de los geles y la incapacidad de las sondas para penetrar en la matriz, las muestras de RNA, separadas por tamaño tras la electroforesis, serán tranferidas a una membrana de Nailon por medio de capilaridad o un sistema de transferencia al vacío. Los sistemas de capilaridad iónica se utilizan pra transferir el ARN desde un gel de electroforesis a una membrana de Northern blot. Una membrana de Nylon cargada positivamente es el soporte mas efectivo para usar en el northern blot ya que los ácidos nucleicos, que están cargados negativamente, tienen una alta afinidad por estas membranas. El buffer de transferencia usado para el ensayo suele contener formamida, dado que ésta consigue disminuir la temperatura de hibridación de la sonda, lo cual previene la degradación del ARN por las altas temperaturas. Una vez que el ARN ha sido transferido a la membrana, este es inmovilizado a través de enlaces covalentes formados con la membrana, lo cual se consigue por medio de luz ultravioleta o calor. Después del marcaje de la sonda, ésta se hibrida con el RNA en la membrana. Las condiciones experimentales que pueden afectar la eficiencia y especificidad de la hibridación están determinadas por las condiciones iónicas, de viscosidad, la presencia de dobles hebras de RNA, bases desemparejadas y composición de las mismas. Finalmente, la membrana debe de lavarse para asegurar que la sonda se ha unido de forma específica y para evitar ruido de fondo en las señales emitidas por la misma. Estas señales serán detectadas por Rayos X y pueden ser cuantificadas mediante técnicas de densitometría. Para crear controles y poder asegurarnos de que no se están mostrando genes que no nos interesen se puede realizar posteriormente la determinación por microarrays o RT-PCR.
TEMA 6-EASTERN BLOT Se considera una extencion de la tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma. pueden examinar la cantidad de proteínas en una muestra y comparar los niveles de presencia entre varios grupos. Otras técnicas ,también usando anticuerpos, permiten la detención de proteínas en tejidos (inmunohistoquímica) y células (inmunocitoquímica). En el Northernblot, se utiliza para identificar las cadenas de ARN de secuencia semejante al ADN que se usa como sonda; a diferencia del tipo Southern que se utiliza para identificar ADN, el método northern sirve para identificar ARN.
TEMA 7 :El Western blot, o inmunoblot, es una técnica analítica usada para detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis en gel se separan las proteínas atendiendo al criterio que se desee: peso molecular, estructura, hidrofobicidad, etc. Hay casi tantas posibilidades como tipos de electroforesis existen. Luego son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella.1 2 Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas. Esta técnica es hoy en día imprescindible en varios campos de la biología, como la biología molecular, la bioquímica, la biotecnología o la inmunología. Existe toda una industria especializada en la venta de anticuerpos (monoclonales y policlonales) contra decenas de miles de proteínas diferentes.3 4 El Western blot fue desarrollado en el laboratorio de George Stark, en la Universidad de Stanford.2 El nombre (Western, occidental en inglés) le fue dado por W. Neal Burnette,5 y consiste en un juego de palabras con una técnica análoga pero que usa DNA, el Southern (sureño) blot, que en este caso debe su nombre a su descubridor, Edwin Southern. Otras técnicas que fueron nombradas siguiendo este criterio son el Northern blot (en el que se separa e identifica RNA), el Eastern blot, el Southwestern blot.
Preparación de la muestra Las muestras pueden ser tomadas de un tejido entero o de un cultivo celular: Una pequeña cantidad del tejido se introduce en un buffer de extracción. Después se procede a homogeneizarlos, sea con una licuadora (para grandes volúmenes de muestra), con un homogenizador (para muestras pequeñas) o por sonicación. Por último se centrifuga para obtener las proteínas en el sobrenadante, la fracción no precipitada.6 Las células pueden lisarse por uno de esos métodos mecánicos. También pueden usarse detergentes, sales o tampones que favorezcan la lisis y solubilicen las proteínas. A veces se añaden inhibidores de proteasas y fosfatasas que evitan la digestión por las enzimas celulares de las proteínas y de las fosfoproteínas, respectivamente.7 Los materiales deben estar a bajas temperaturas (~4 °C) para evitar la desnaturalización proteica. Para separar proteínas de compartimentos y orgánulos celulares es preciso combinar técnicas mecánicas - como filtraciones y centrifugaciones - y bioquímicas.
TEMA 9:LA TÉCNICA DE TRANSFERENCIA BLOTTING La técnica de transferencia (blotting) se basa en el mismo fundamento que la autorradiografía, y permite identificar la presencia de una banda concreta en un gel de electroforesis, bien sea de proteínas o de ácidos nucleicos: Si se trata de proteínas, la técnica se llama Western blot. Se utilizan anticuerpos marcados con radioactividad para identificar cuál es la banda que presenta la proteína que me interesa localizar Si se trata de DNA, la técnica se llama Southern blot. Se utilizan sondas de DNA (moléculas de DNA marcadas con radioactividad y con una secuencia conocida) para identificar qué bandas del gel contienen secuencias complementarias a la de la sonda Si se trata de RNA, la técnica se llama Northern blot. Se utilizan sondas de RNA (moléculas de RNA marcadas con radioactividad y con una secuencia conocida) para identificar qué bandas del gel contienen secuencias complementarias a la de la sonda
TEMA10: ELECTROFORESIS DE PROTEÍNAS EN SUERO Este examen mide los tipos de proteína en la parte líquida (suero) de una muestra de sangre. Ver también: Forma en que se realiza el examen Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción
La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína. La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL). PREPARACIÓN PARA EL EXAMEN A usted le pueden solicitar no comer ni beber nada durante 12 horas antes de un examen de electroforesis de lipoproteínas. El médico le puede solicitar que deje de tomar medicamentos que pudieran afectar el examen. NO suspenda ningún medicamento sin consultarlo previamente con éste.
Los medicamentos que pueden afectar la medición de las proteínas totales incluyen clorpromazina, corticoesteroides, isoniazida, neomicina, fenacemida, salicilatos, sulfamidas y tolbutamida.
LO QUE SE SIENTE DURANTE EL EXAMEN Cuando se inserta la aguja para extraer la sangre, algunas personas sienten un dolor moderado, mientras que otras sólo sienten un pinchazo o sensación de picadura. Posteriormente, puede haber algo de sensación pulsátil. RAZONES POR LAS QUE SE REALIZA EL EXAMEN Las proteínas están compuestas de aminoácidos y son componentes importantes de todas las células y los tejidos. Existen muchas clases de proteínas en el cuerpo, con muchas funciones diferentes. Los ejemplos de proteínas son: las enzimas, algunas hormonas, la hemoglobina, la lipoproteína de baja densidad (colesterol "malo") y otras. Las proteínas séricas se clasifican como albúmina y globulinas. La albúmina es la proteína de mayor concentración en el suero. Trasporta muchas moléculas pequeñas, pero también es importante para impedir que el líquido se escape de los vasos sanguíneos hacia los tejidos. Las globulinas se dividen en alfa-1, alfa-2, beta y gammaglobulinas. En general, los niveles de las proteínas alfa y gammaglobulinas aumentan cuando hay inflamación en el cuerpo.
2012-1330 Tema 2 :Reacción de cadena polimerasa (PCR) La reacción en cadena de la polimerasa, ya más conocida como PCR, es una técnica que permite replicar entre cientos de miles y millones de veces, en el transcurrir de pocas horas e in vitro, pequeñas cantidades de ADN. Uno de los aportes fundamentales de esta metodología a la investigación básica en biología molecular consiste, precisamente, en la rapidez y eficiencia mediante las cuales se realiza una tarea que antes requería largas y tediosas jornadas. El producto quese obtiene al finalizar la reacción es una gran cantidad de un fragmento génico con alto grado de pureza- favorece la tarea de los investigadores empeñados en ampliar nuestros conocimientos sobre la estructura y función de los genes. El método se basa, en su forma más simple en la realización de tres reacciones sucesivas llevadas a cabo a distintas temperaturas. Estas reacciones se repiten cíclicamente entre veinte y cuarenta veces (véase la figura 1). La muestra se calienta, en el primer paso, hasta lograr la separación de las dos cadenas que constituyen el ADN, hecho que se conoce como "desnaturalización". En el segundo paso, la temperatura se reduce para permitir el "apareamiento" de cada una de dos cadenas cortas de nucleótidos (oligonucleóridos) con cada una de las hebras separadas del ADN molde. Se trata de segmentos de ADN de cadena simple, sintetizados en el laboratorio y diseñados de manera tal que permiten definir los límites del tramo de ADN que se desea replicar. Obviamente, para que se pueda producir el apareamiento, cada uno de estos oligonucleótidos, a los que se denomina "iniciadores" o primers, debe ser complementario del tramo al que tienen que unirse en las cadenas separadas del ADN molde. En tercer lugar, una enzima ADN polimerasa extiende los primers, en el espacio comprendido entre ambos, sintetizando las secuencias complementarias de las hebras del ADN molde. Para ello, la ADN polimerasa usa desoxidonucleósidos trifosfato agregados a la mezcla de reacción. La temperatura a la que se realiza el tercer paso está condicionada por aquélla a la cual "trabaja" la enzima ADN polimerasa. Al cabo del primer ciclo de tres reacciones (desnaturalización, apareamiento, extensión) el tramo de ADN elegido se ha duplicado y el doble de su cantidad original se encuentra disponible para ser nuevamente replicado en un segundo ciclo. El resultado de la aplicación de numerosos ciclos "en cadena" da lugar a la amplificación geométrica del segmento de ADN delimitado por los primers.Para replicar el ADN, la técnica PCR hizo uso, en un principio, de la ADN polimerasa de la bacteria Escherichia coli. Pero esta enzima resulta desactivada debido a la alta temperatura requerida para desnaturalizarla doble cadena del ADN, por lo cual debía agregarse enzima fresca al comenzar el tercer paso de cada ciclo. Este inconveniente fue solucionado de manera ingeniosa cuando se la reemplazó por su equivalente de la bacteria "termófila" Thermus aquaticus. En efecto, la ADN polimerasa de este microrganismo, denominada Taq polimerasa, actúa eficientemente entre los 75° C y los 80° C y resiste más de dos horas a 93° C. De esta manera es posible mezclar todos los componentes de la PCR al comenzar el primer ciclo y la reacción en cadena puede llevarse a cabo mediante equipos automatizados que realizarán los ciclos térmicos programados.
2012-1330 tema 3: Enzimas de restrincion (BRENDA) Las enzimas de restricción son proteínas cuya función es cortar las hebras de ADN. Se podría decir que son “tijeras moleculares” que cortan ADN. Lo hacen en forma específica. Esto significa que cada enzima reconoce un sitio particular del ADN, es decir que reconoce una secuencia particular de nucleótidos. Esa secuencia específica para cada enzima se denomina “sitio de restricción”. Una vez que la enzima reconoce estos sitios, se posiciona sobre la molécula de ADN y corta dentro o en torno de esa secuencia. Las enzimas de restricción, conocidas también como endonucleasas, sólo cortan el ADN si reconocen en su interior una secuencia específica de nucleótidos. Estas enzimas fueron descubiertas en microorganismos. De hecho, se encuentran sólo en organismos procariotas (bacterias). Por esto, se les dio una nomenclatura asociada al organismo de donde provienen. Por ejemplo, las enzimas de restricción que se descubrieron en la bacteria Escherichia coli se denominan Eco. Existen diferentes tipos de enzimas Eco que se diferencian en la secuencia que reconocen y cortan. Para diferenciarlas se les agregan letras y números romanos, por ejemplo: Eco RI (Eco “erre” “uno”). Así, el sitio de restricción para EcoRI es la secuencia GAATTC, como muestra la ilustración anterior. Una vez que la enzima encontró ese sitio en el ADN, se acerca a la hebra de ADN y realiza el corte entre la G y la A. Al investigar otras especies de bacterias se descubrieron cientos de enzimas de restricción distintas y cada una reconoce una región específica. Estas enzimas fueron descubiertas en la década del ’70 y hasta la fecha existen más de 250 enzimas de restricción. Se cree que la función natural de estas enzimas en las bacterias es protegerlas contra ADN de virus que podrían ingresar en sus células. De esta manera, la bacteria utiliza estas “tijeras moleculares” para cortar en pedacitos el ADN viral que la infecta. El ADN propio de la bacteria no se corta, pues lo tiene “protegido” contra sus propias enzimas de restricción. Usos de las enzimas de restricción Las enzimas de restricción tienen diferentes aplicaciones que son de gran importancia en investigaciones en biología molecular y en las técnicas que emplea la biotecnología moderna: 1. Hacer mapa de restricción de un plásmido o bacteriófago. El ADN se corta con varias enzimas de restricción, solas y en parejas, para determinar el número de sitios de corte y sus posiciones relativas en la molécula, el orden y la distancia entre ellos. 2. Fragmentar ADN para separación por electroforesis. Los fragmentos obtenidos después de la actuación de las distintas enzimas de restricción, se pueden separar por tamaños mediante la técnica de electroforesis y así estudiar los distintos fragmentos. Por ejemplo: para la técnica de Southern blotting o en las usadas para identificar polimorfismos de ADN en distintos individuos . 3. Generación de fragmentos para ser clonados en los vectores apropiados, y crear ADN recombinante. Se puede cortar una molécula de ADN con una enzima y, con el mismo tipo de enzima, cortar el fragmento de ADN de interés para clonar. Se unen con ligasas estas dos moléculas de ADN, generando así una molécula de ADN recombinante. Este vector recombinante puede usarse para transformar células que expresen el gen de interés clonado (si se usó un vector de expresión con el promotor adecuado) o puede usarse simplemente para tener clonado (“guardado”) ese fragmento de ADN de interés. Por ejemplo: para los proyectos de secuenciación de genomas
2012-1330 tema 4: ELISA directo (Sandwish) La técnica de ELISA (Enzyme Linked InmunoSorbent Assay) es un procedimiento de ensayo inmuno enzimático. Se basa en la detección antígeno(Ag) o anticuerpo (Ac), La técnica de ELISA es un ensayo inmunoenzimático ampliamente empleado en el área médica para la cuantificación de moléculas especialmente de aquellas que experimentan cambios en diferentes estados como pueden ser infecciones por bacterias, virus, hongos o parásitos o fases activas de enfermedades autoinmunes. Como ejemplo se pueden medir por ELISA hormonas, autoanticuerpos, inmunoglobulinas contra antígenos de patógenos, toxinas, antígenos. Las técnicas de ELISA son también muy usadas en los ensayos de nuevos medicamentos para comprobar sus efectos cuantificando las moléculas de interés en cada caso. Existen numerosas variantes de este tipo de ensayo. Entre ellos se encuentran los métodos directo e indirecto. El método directo permite la detección del antígeno con un anticuerpo específico conjugado con una enzima como sistema de marcaje. Técnica: Las placas ELISA se preparan recubriendo los pocillos con las soluciones en las que se sospecha se encuentra el antígeno. Se incuban con anticuerpos marcados. Indican la presencia de antígeno en la solución analizada. Es necesario incluir controles negativos que serán muestras del mismo tipo de las analizadas (sangre, orina) pero en las que se tenga la certeza de la ausencia del antígeno buscado. Asimismo se incluyen controles positivos (soluciones donde se encuentra el antígeno buscado). Elisa sandwish Se trata de un ensayo muy empleado en el que se recubre el pocillo con un primer anticuerpo anti-antígeno. Después de lavar el exceso de anticuerpo se aplica la muestra problema en la que se encuentra el antígeno, que será retenido en el pocillo al ser reconocido por el primer anticuerpo. Después de un segundo lavado que elimina el material no retenido se aplica una solución con un segundo anticuerpo anti-antígeno marcado. Así pues cada molécula de antígeno estará unida a un anticuerpo en la base que lo retiene y un segundo anticuerpo, al menos, que lo marca. Este ensayo tiene una gran especificidad y sensibilidad debido a la amplificación de señal que permite el segundo anticuerpo.
2012-1330 tema 5:ELISA Indirecto En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA. Es el ensayo más popular, como lo es la inmunofluorescencia indirecta, pues un mismo secundario marcado y un mismo sistema enzimático permite cuantificar una gran variedad de antígenos, por eso es un método más polivalente y barato, aunque se pierda algo de precisión por tener un eslabón más con respecto al método directo. La dilución de la solución que contiene el anticuerpo primario (por ejemplo: suero sanguíneo) es un factor muy importante a tener en cuenta para evitar la aparición de falsos negativos, ya que si la muestra está muy diluida nosaldrá positiva si la titulación de anticuerpos es muy baja. Es decir, aunque los anticuerpos están presentes, la prueba no da positivo porque la concentración de anticuerpos específicos contra el antígeno que está pegado en el fondo del pocillo no es suficiente como para dar una señal detectable.El ELISA indirecto es el método de elección para detectar la presencia de anticuerpos séricos contra el virus de la inmunodeficiencia humana (VIH), agente causante del síndrome de inmunodeficiencia adquirida (SIDA). Según esta técnica, proteínas recombinantes de la envoltura y el núcleo del VIH se absorben como antígenos en fase sólida a los pocillos. Las personas afectadas de VIH producen anticuerpos séricos contra epítopos en estas proteínas víricas. En general, el ELISA indirecto permite detectar anticuepos séricos contra VIH desde las primeras seis semanas de una infección.
2012-1330 tema 6:Tecnicas Blotting El término “blotting” hace referencia a la transferencia de macromoléculas biológicas desde un gel hasta una membrana y su posterior detección en la superficie de la misma. Este proceso es necesario ya que todas estas macromoléculas están embebidas dentro de la matriz que forma el gel, lo que imposibilita realizar su detección, mientras que en la membrana, se encuentran accesibles al estar adheridas sobre la superficie de la misma. La técnica del Western Blot (también llamado “inmunoblotting” debido a que se utilizan anticuerpos para detectar el antígeno o los antígenos específicos de interés) fue descrita por primera vez por Towbin et al. en 1979 y en la actualidad es una técnica de rutina en todos los laboratorios que realizan análisis de proteínas. La especificidad de la unión antígeno – anticuerpo permite la detección de una única proteína dentro de una mezcla compleja de otras proteínas. En la actualidad se utiliza como criterio de identificación positivo de una proteína especifica en una mezcla compleja y para obtener datos cualitativos y semicuantitativos sobre la misma. El primer paso de todo Western Blot es la separación de las macromoléculas mediante geles de electroforesis; después de la misma, las macromoléculas ya separadas en función de su diferente peso molecular se transfieren a una segunda matriz, generalmente una membrana de nitrocelulosa o PVDF. Posteriormente, se bloquea la membrana para evitar la unión inespecífica a su superficie de los anticuerpos que se van a utilizar para la detección de la proteína de interés. En el siguiente paso, se une a dicha proteína transferida un anticuerpo especifico marcado con una enzima y, finalmente, se añade un sustrato apropiado para dicha enzima con lo que se produce un producto detectable como, por ejemplo, un precipitado cromogénico o fluorogénico en la membrana. En la actualidad, los métodos más sensibles emplean sustratos quimioluminiscentes que cuando se combinan con la enzima correspondiente, producen luz como producto final, que puede detectarse mediante una película o una cámara CCD. En todos los casos y sea cual sea el sustrato que se utilice, la intensidad de la señal se correlaciona con la cantidad del antígeno en la superficie de la membrana.
2012-1330 tema 7:Electroforesis de proteínas Es un método de separación de proteínas mediante la aplicación de un campo eléctrico. Existen diferentes tipos en función del tipo de separación empleado: electroforesis de zona (separación en función de la carga), isoeléctrico enfoque y separación por tamaño en tamiz molecular (también aplicable a ácidos nucleicos). Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina).Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (-) o cátodo (+) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración. Isoelectroenfoque: en lugar de separar las proteínas en función de su carga a un pH dado, se separan en función de su punto isoeléctrico (pI): el pI es el pH en el que la carga neta de la proteína es nula, y depende de la composición aminoacídica de la proteína. Se crea un gradiente de pH mediante anfolitos (estabilizan el pH a lo largo del gel). Cada proteína migrará hasta alcanzar su pI, punto en el cual precipitará al acumularse (de ahí el nombre, isoelectroenfoque).También se suelen utilizar acoplados a zimogramas si deseamos determinar el pI de una enzima o en electroforesis bidimensional como primera dimension. Separación por tamaño: permite separar proteínas y ácidos nucleicos. En el caso de las proteínas, deben ser tratadas con SDS (sodio dodecil sulfato) para que su carga sea negativa y todas migren hacia el ánodo (no es necesario hacer eso con los ácidos nucleicos, ya que tienen carga negativa); la separación se hace en medios de soporte en el que se ha creado un tamiz molecular (matriz), que hace que las proteínas más pequeñas corran más que las más grandes.Una vez separadas las proteínas, deben fijarse y teñirse para poder ser visualizadas. Hay diferentes protocolos en función de lo que se quiere estudiar: fijación por calor o química y tinción en caso de estudios no específicos (proteinograma y electroforesis Hb, por ejemplo) o fijación mediante anticuerpos previa a la tinción en caso de estudiar proteínas específicas (inmunofijación).Por lo general para la tinción de geles de poliacrilamida se utiliza tinción con plata, tinción de Coomassie o tinción fluorescente. Dependiendo del fin de nuestro análisis. La tinción con plata no es utilizada si se desean hacer análisis por espectrometría de masas (MS) pero es más sensible que la tinción con Azul de Coomassie, por otro lado, la tinción fluorescente es muy sensible y compatible con MS pero los costos de tinción son elevados.Una tinción sensible y barata es la denominada "Blue Silver" o Coomassie G250.1 Además, esta tinción es compatible con MS. Está compuesta por azul de coomassie diluido en una solución coloidal y se utiliza agua destilada para destiñir el gel de poliacrilamida.
2012-1330 tema 8:El ADN mitocondrial Es un fragmento de ácido nucleico encontrado dentro de las mitocondrias. Las mitocondrias son pequeños organelos o estructuras celulares que se encuentran dentro de las células eucarióticas (generalmente las células animales). Estos organelos se usan básicamente para la producción y generación de energía para que la célula lleve a cabo sus funciones. Las plantas tienen cloroplastos. Las células animales tienen mitocondrias. Y cada una de estas mitocondrias tiene su propio genoma. Su propia molécula circular de ADN. Y eso es el ADN mitocondrial, porque en realidad es diferente del ADN que se encuentra en el núcleo de las células. No se encuentra dentro del núcleo. El número de genes en el ADN mitocondrial es de 37, frente a los 20.000 - 25.000 genes del ADN cromosómico nuclear humanos.Codifica dos ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa. El ADN mitocondrial está en replicación constante, independientemente del ciclo y del tipo celular. Se piensa que tiene lugar de forma asíncrona, es decir, que tiene lugar en las dos cadenas en tiempos diferentes y con dos orígenes distintos hacia direcciones contrarias. El comienzo tendría lugar en el origen de la cadena pesada, situado en el bucle D, y replicaría ésta tomando como molde la cadena ligera. Cuando se alcanza el segundo origen, situado a dos tercios de distancia del primero, comienza la segunda ronda de replicación en sentido opuesto. Se ha propuesto un nuevo sistema de replicación que coexistiría con el primero. Sería bidireccional y comportaría una coordinación entre hebras directas y retrasadas. Tradicionalmente se ha considerado que el ADN mitocondrial humano se hereda sólo por vía materna. Según esta concepción, cuando un espermatozoide fecunda un óvulopenetra el núcleo y su cola junto con sus mitocondrias son destruidos en el óvulo materno. Por lo tanto, en el desarrollo del cigoto sólo intervendrían las mitocondrias contenidas en el óvulo. Sin embargo, se ha demostrado que las mitocondrias del espermatozoide pueden ingresar al óvulo. Según algunos autores el ADN mitocondrial del padre puede perdurar en algunos tejidos, como los músculos.Según otros, no llega a heredarse al ser marcado por ubiquitinación y degradado. El ADN mitocondrial puede ser usado para identificar individuos junto con otra evidencia. También es usado por laboratorios forenses para caracterizar viejas muestras de esqueleto humano. Distinto que el ADN nuclear, el ADN mitocondrial no sirve para identificar individuos sin ambigüedad, pero si para detectar parentescos entre grupos de individuos; es usado entonces para comparaciones entre personas desaparecidas y restos no identificados y sus familiares. El ADN mitocondrial humano tiene características únicas que lo hacen muy apropiado para estudios microevolutivos: la herencia del genoma mitocondrial se realiza exclusivamente por la vía materna, sin recombinarse; hay un fragmento en este genoma de 400pb (pares de bases) altamente polimórfico, y posee una alta frecuencia de mutaciones (5 a 10 veces mayor que el ADN nuclear)10 Este ADN se puede extraer de muestras de cualquier tejido, incluso de la sangre y del tejido óseo. Gracias a su presencia en el hueso se puede obtener el genoma de individuos ya muertos desde hace muchos años. El análisis de la secuencia genómica se usa para estudiar las relaciones filogenéticas, no sólo en humanos sino, también en muchos otros organismos. Por este motivo se utiliza para determinar variabilidad en poblaciones naturales (para ver si hay o no endogamia), información útil para la conservación de especies en peligro de extinción.
2012-1330 tema 9;Anticuerpos monoclonales Son anticuerpos homogéneos producido por una célula híbrida producto de la fusión de un clon de linfocitos B descendiente de una sola y únicacélula madre y una célula plasmática tumoral. Los anticuerpos monoclonales mAB, de la frase en inglés con idéntico significado que en español: monoclonal antibody, son anticuerpos idénticos porque son producidos por un solo tipo de célula del sistema inmune, es decir, todos los clones proceden de una sola célula madre. Es posible producir anticuerpos monoclonales que se unan específicamente con cualquier molécula con carácter antigénico. Este fenómeno es de gran utilidad en bioquímica, biología molecular y medicina. Si una sustancia extraña (antígeno) se inyecta en el cuerpo de un ratón o un humano, alguna de las células B de su sistema inmune se transformarán en células plasmáticasy empezarán a producir anticuerpos que se unirán a ese antígeno. Cada célula B produce un solo tipo de anticuerpo, pero diferentes linfocitos B producirán anticuerpos estructuralmente diferentes que se unen a distintas partes del antígeno. Esta mezcla fisiológica natural de anticuerpos es conocida como Antisuero policlonal.Para producir anticuerpos monoclonales, primero se extraen células B del bazo de un animal que ha sido expuesto al antígeno. Estas células B son fusionadas en presencia de PEG (polietilenglicol) con células tumorales de mieloma múltiple (un tipo de cáncer) que pueden crecer indefinidamente en cultivo celular. Esta fusión hace a las membranas celulares más permeables. Estas células fusionadas híbridas, llamadas hibridomas pueden multiplicarse rápida e indefinidamente, puesto que son células tumorales después de todo y pueden producir gran cantidad de anticuerpos. Los hibridomas son suficientemente diluidos y cultivados para obtener un número diferente de determinadas colonias, las cuales producen sólo un tipo de anticuerpo. Los anticuerpos de diferentes colonias son analizados para conocer su capacidad de unirse a un antígeno determinado, por ejemplo con un tipo de test llamado ELISA, y para seleccionarse y aislarse de la manera más efectiva. Nuestro sistema inmune reconoce y ataca elementos ajenos al organismo, como bacterias causantes de enfermedades, virus y otros agentes infecciosos. Este sistema de defensa está basado en células especializadas y proteínas que protegen el organismo. Estas proteínas especializadas incluyen anticuerpos y nuestro organismo produce una enorme variedad de anticuerpos para ser capaz de interaccionar con prácticamente todo posible patógeno. Los anticuerpos tienen dos características muy útiles. En primer lugar, son extremadamente específicos, es decir, cada anticuerpo se une y ataca un único antígeno. En segundo lugar, algunos anticuerpos, una vez activados por la presencia de la enfermedad, continúan confiriendo resistencia contra esa enfermedad; ejemplos clásicos son los anticuerpos de las enfermedades de la infancia.
Podriamos decir que la La transcripción del ADN es el primer proceso de la expresión génica, mediante el cual se transfiere la información contenida en la secuencia del ADN hacia la secuencia de proteína utilizando diversos ARN como intermediarios. Durante la transcripción genética, las secuencias de ADN son copiadas a ARN mediante una enzima llamada ARN polimerasa que sintetiza un ARN mensajero que mantiene la información de la secuencia del ADN. De esta manera, la transcripción del ADN también podría llamarse síntesis del ARN mensajero.
TRADUCCION : La traducción es el segundo proceso de la síntesis proteica (parte del proceso general de la expresión génica). La traducción ocurre tanto en el citoplasma, donde se encuentran los ribosomas, como también en el retículo endoplasmático rugoso (RER). Los ribosomas están formados por una subunidad pequeña y una grande que rodean al ARNm. En la traducción, el ARN mensajero se decodifica para producir un polipéptido específico de acuerdo con las reglas especificadas por el código genético. Es el proceso que convierte una secuencia de ARNm en una cadena de aminoácidos para formar una proteína. Es necesario que la traducción venga precedida de un proceso de transcripción. El proceso de traducción tiene cuatro fases: activación, iniciación, elongación y terminación (entre todos describen el crecimiento de la cadena de aminoácidos, o polipéptido, que es el producto de la traducción). En la activación, el aminoácido (AA) correcto se une al ARN de transferencia (ARNt) correcto. Aunque técnicamente esto no es un paso de la traducción, es necesario para que se produzca la traducción. El AA se une por su grupo carboxilo con el OH 3' del ARNt mediante un enlace de tipo éster. Cuando el ARNt está enlazado con un aminoácido, se dice que está "cargado". La iniciación supone que la subunidad pequeña del ribosoma se enlaza con el extremo 5' del ARNm con la ayuda de factores de iniciación (FI), otras proteínas que asisten el proceso. La elongación ocurre cuando el siguiente aminoacil-ARNt (el ARNt cargado) de la secuencia se enlaza con el ribosoma además de con un GTP y un factor de elongación. La terminación del polipéptido sucede cuando la zona A del ribosoma se encuentra con un codón de parada (sin sentido), que son el UAA, UAG o UGA. Cuando esto sucede, ningún ARNt puede reconocerlo, pero el factor de liberación puede reconocer los codones sin sentido y provoca la liberación de la cadena polipeptídica. La capacidad de desactivar o inhibir la traducción de la biosíntesis de proteínas se utiliza en antibióticos como la anisomicina, la cicloheximida, el cloranfenicol y la tetraciclina.
Yuri solis 2012-1313 PCR El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Desnaturalización Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación, Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario.
La tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma.
S outhern Blot para adn y proteina El Southern Blot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizando sonicación ó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAds ó RNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.El traspaso de las muestras desde el gel al filtro es una etapa esencial, porque el reconocimiento debases complementarias entre sonda y secuencias de DNA en el gel es muy ineficiente.Cabe señalar que el nombre de la técnica Southern Blot deriva en parte del apellido Southern delinvestigador que la desarrolló y de la palabra en inglés Blot que significa traspasar.
KERY JOUSSETT SANCHEZ ALVAREZ 2012-0919 TIPOS DE ARN: El ARN mensajero (ARNm) es el tipo de ARN que lleva la información del ADN a los ribosomas, el lugar de la síntesis de proteínas. La secuencia de nucleótidos del ARNm determina la secuencia de aminoácidos de la proteína.21 Por ello, el ARNm es denominado ARN codificante. No obstante, muchos ARN no codifican proteínas, y reciben el nombre de ARN no codificantes; se originan a partir de genes propios (genes ARN), o son los intrones rechazados durante el proceso de splicing. Son ARN no codificantes el ARN de transferencia (ARNt) y el ARN ribosómico (ARNr), que son elementos fundamentales en el proceso de traducción, y diversos tipos de ARN reguladores.22 Ciertos ARN no codificantes, denominados ribozimas, son capaces de catalizar reacciones químicas como cortar y unir otras moléculas de ARN,23 o formar enlaces peptídicos entre aminoácidos en el ribosoma durante la síntesis de proteínas.24 ARN implicados en la síntesis de proteínas. Ribosoma 50S mostrando el ARNr (amarillo), las proteínas (azul) y el centro activo, la adenina 2486 (rojo). ARN mensajero El ARN mensajero (ARNm o RNAm) lleva la información sobre la secuencia de aminoácidos de la proteína desde el ADN, lugar en que está inscrita, hasta el ribosoma, lugar en que se sintetizan las proteínas de la célula. Es, por tanto, una molécula intermediaria entre el ADN y la proteína y apelativo de "mensajero" es del todo descriptivo. En eucariotas, el ARNm se sintetiza en el nucleoplasma del núcleo celular y donde es procesado antes de acceder al citosol, donde se hallan los ribosomas, a través de los poros de la envoltura nuclear. ARN de transferencia[editar · editar código] Los ARN de transferencia (ARNt o tRNA) son cortos polímeros de unos 80 nucleótidos que transfiere un aminoácido específico al polipéptido en crecimiento; se unen a lugares específicos del ribosoma durante la traducción. Tienen un sitio específico para la fijación del aminoácido (extremo 3') y un anticodón formado por un triplete de nucleótidos que se une al codón complementario del ARNm mediante puentes de hidrógeno.22 ARN ribosómico o ribosomal[editar · editar código] El ARN ribosomico o ribosomal (ARNr o RNAr) se halla combinado con proteínas para formar los ribosomas, donde representa unas 2/3 partes de los mismos. En procariotas, la subunidad mayor del ribosoma contiene dos moléculas de ARNr y la subunidad menor, una. En los eucariotas, la subunidad mayor contiene tres moléculas de ARNr y la menor, una. En ambos casos, sobre el armazón constituido por los ARNr se asocian proteínas específicas. El ARNr es muy abundante y representa el 80% del ARN hallado en el citoplasma de las células eucariotas.25 Los ARN ribosómicos son el componente catalítico de los ribosomas; se encargan de crear los enlaces peptídicos entre los aminoácidos del polipéptido en formación durante la síntesis de proteínas; actúan, pues, como ribozimas. ARN reguladores[editar · editar código] Muchos tipos de ARN regulan la expresión génica gracias a que son complementarios de regiones específicas del ARNm o de genes del ADN. ARN de interferencia[editar · editar código] Los ARN interferentes (ARNi o iRNA) son moléculas de ARN que suprimen la expresión de genes específicos mediante mecanismos conocidos globalmente como ribointerferencia o interferencia por ARN. Los ARN interferentes son moléculas pequeñas (de 20 a 25 nucléotidos) que se generan por fragmentación de precursores más largos. Se pueden clasificar en tres grandes grupos:26
ARN El ácido ribonucleico (ARN o RNA) es un ácido nucleico formado por una cadena de ribonucleótidos. Está presente tanto en las células procariotas como en las eucariotas, y es el único material genético de ciertos virus (virus ARN). El ARN celular es lineal y de hebra sencilla, pero en el genoma de algunos virus es de doble hebra. En los organismos celulares desempeña diversas funciones. Es la molécula que dirige las etapas intermedias de la síntesis proteica; el ADN no puede actuar solo, y se vale del ARN para transferir esta información vital durante la síntesis de proteínas (producción de las proteínas que necesita la célula para sus actividades y su desarrollo). Varios tipos de ARN regulan la expresión génica, mientras que otros tienen actividad catalítica. El ARN es, pues, mucho más versátil que el ADN.
ADN El ácido desoxirribonucleico, abreviado como ADN, es un ácido nucleico que contiene instrucciones genéticas usadas en el desarrollo y funcionamiento de todos los organismos vivos conocidos y algunos virus, y es responsable de su transmisión hereditaria. El papel principal de la molécula de ADN es el almacenamiento a largo plazo de información. Muchas veces, el ADN es comparado con un plano o una receta, o un código, ya que contiene las instrucciones necesarias para construir otros componentes de las células, como las proteínas y las moléculas de ARN. Los segmentos de ADN que llevan esta información genética son llamados genes, pero las otras secuencias de ADN tienen propósitos estructurales o toman parte en la regulación del uso de esta información genética. Desde el punto de vista químico, el ADN es un polímero de nucleótidos, es decir, un polinucleótido. Un polímero es un compuesto formado por muchas unidades simples conectadas entre sí, como si fuera un largo tren formado por vagones. En el ADN, cada vagón es un nucleótido, y cada nucleótido, a su vez, está formado por un azúcar (la desoxirribosa), una base nitrogenada (que puede ser adenina→A, timina→T, citosina→C o guanina→G) y un grupo fosfato que actúa como enganche de cada vagón con el siguiente. Lo que distingue a un vagón (nucleótido) de otro es, entonces, la base nitrogenada, y por ello la secuencia del ADN se especifica nombrando sólo la secuencia de sus bases. La disposición secuencial de estas cuatro bases a lo largo de la cadena (el ordenamiento de los cuatro tipos de vagones a lo largo de todo el tren) es la que codifica la información genética: por ejemplo, una secuencia de ADN puede ser ATGCTAGATCGC... En los organismos vivos, el ADN se presenta como una doble cadena de nucleótidos, en la que las dos hebras están unidas entre sí por unas conexiones denominadas puentes de hidrógeno.
Compuesto orgánico o molécula orgánica es una sustancia química que contiene carbono, formando enlaces carbono-carbono y carbono-hidrógeno. En muchos casos contienen oxígeno, nitrógeno, azufre, fósforo, boro, halógenos y otros elementos menos frecuentes en su estado natural. Estos compuestos se denominan moléculas orgánicas. Algunos compuestos del carbono, carburos, los carbonatos y los óxidos de carbono, no son moléculas orgánicas. La principal característica de estas sustancias es que arden y pueden ser quemadas (son compuestos combustibles). La mayoría de los compuestos orgánicos se producen de forma artificial mediante síntesis química aunque algunos todavía se extraen de fuentes naturales. Las moléculas orgánicas pueden ser de dos tipos: Moléculas orgánicas naturales: son las sintetizadas por los seres vivos, y se llaman biomoléculas, las cuales son estudiadas por la bioquímica y las derivadas del petróleo como los hidrocarburos. Moléculas orgánicas artificiales: son sustancias que no existen en la naturaleza y han sido fabricadas o sintetizadas por el hombre, por ejemplo los plásticos. La línea que divide las moléculas orgánicas de las inorgánicas ha originado polémicas e históricamente ha sido arbitraria, pero generalmente, los compuestos orgánicos tienen carbono con enlaces de hidrógeno, y los compuestos inorgánicos, no. Así el ácido carbónico es inorgánico, mientras que el ácido fórmico, el primer ácido carboxilico, es orgánico. El anhídrido carbónico y el monóxido de carbono, son compuestos inorgánicos. Por lo tanto, todas las moléculas orgánicas contienen carbono, pero no todas las moléculas que contienen carbono son moléculas orgánicas.
Se denomina compuesto químico inorgánico a todos aquellos compuestos que están formados por distintos elementos, pero en los que su componente principal no siempre es el carbono, siendo el agua el más abundante. En los compuestos inorgánicos se podría decir que participan casi la totalidad de elementos conocidos. como el oro,plata . Mientras que un compuesto orgánico se forma de manera natural tanto en animales como en vegetales, uno inorgánico se forma de manera ordinaria por la acción de varios fenómenos físicos y químicos: electrólisis, fusión, etc. También podrían considerarse agentes de la creación de estas sustancias a la energía solar, el agua, el oxígeno. Los enlaces que forman los compuestos inorgánicos suelen ser iónicos o covalentes. Ejemplos de compuestos inorgánicos: Cada molécula de cloruro de sodio (NaCl) está compuesta por un átomo de sodio y otro de cloro. Cada molécula de agua (H2O) está compuesta por dos átomos de hidrógeno y uno de oxígeno. Cada molécula de amoníaco (NH3) está compuesta por un átomo de nitrógeno y tres de hidrógeno. El anhídrido carbónico se encuentra en la atmósfera en estado gaseoso y los seres vivos aerobios lo liberan hacia ella al realizar la respiración. Su fórmula química, CO2, indica que cada molécula de este compuesto está formada por un átomo de carbono y dos de oxígeno. El CO2 es utilizado por algunos seres vivos autótrofos como las plantas en el proceso de fotosíntesis para fabricar glucosa. Aunque el CO2 contiene carbono, no se considera como un compuesto orgánico porque no contiene hidrógeno.
El descubrimiento de los ácidos nucleicos se debe a Friedrich Miescher, quien en el año 1869 aisló de los núcleos de las células una sustancia ácida a la que llamó nucleína,1 nombre que posteriormente se cambió a ácido nucleico. Posteriormente, en 1953, James Watson y Francis Crick descubrieron la estructura del ADN, empleando la técnica de difracción de rayos X. Los ácidos nucleicos son grandes polímeros formados por la repetición de monómeros denominados nucleótidos, unidos mediante enlaces fosfodiéster. Se forman, así, largas cadenas; algunas moléculas de ácidos nucleicos llegan a alcanzar tamaños gigantescos, con millones de nucleótidos encadenados. Los ácidos nucleicos almacenan la información genética de los organismos vivos y son los responsables de la transmisión hereditaria. Existen dos tipos básicos, el ADN y el ARN.
Formula de los acidos nucleicos X+Y- xy X y + pn =
TIPOS
Existen dos tipos de ácidos nucleicos: ADN (ácido desoxirribonucleico) y ARN (ácido ribonucleico), que se diferencian: por el glúcido (la pentosa es diferente en cada uno; ribosa en el ARN y desoxirribosa en el ADN); por las bases nitrogenadas: adenina, guanina, citosina y timina, en el ADN; adenina, guanina, citosina y uracilo, en el ARN; en la inmensa mayoría de organismos, el ADN es bicatenario (dos cadenas unidas formando una doble hélice), mientras que el ARN es monocatenario (una sola cadena), aunque puede presentarse en forma extendida, como el ARNm, o en forma plegada, como el ARNt y el ARNr; en la masa molecular: la del ADN es generalmente mayor que la del ARN.
Las proteínas son moléculas formadas por cadenas lineales de aminoácidos. El término proteína proviene de la palabra francesa protéine y ésta del griego πρωτεῖος (proteios), que significa 'prominente, de primera calidad'.1 Por sus propiedades físico-químicas, las proteínas se pueden clasificar en proteínas simples (holoproteidos), que por hidrólisis dan solo aminoácidos o sus derivados; proteínas conjugadas (heteroproteidos), que por hidrólisis dan aminoácidos acompañados de sustancias diversas, y proteínas derivadas, sustancias formadas por desnaturalización y desdoblamiento de las anteriores. Las proteínas son necesarias para la vida, sobre todo por su función plástica (constituyen el 80% del protoplasma deshidratado de toda célula), pero también por sus funciones biorreguladoras (forman parte de las enzimas) y de defensa (los anticuerpos son proteínas).2 Las proteínas desempeñan un papel fundamental para la vida y son las biomoléculas más versátiles y diversas. Son imprescindibles para el crecimiento del organismo y realizan una enorme cantidad de funciones diferentes, entre las que destacan: Estructural. Esta es la función más importante de una molecula (Ej: colágeno), Inmunológica (anticuerpos), Enzimática (Ej: sacarasa y pepsina), Contráctil (actina y miosina). Homeostática: colaboran en el mantenimiento del pH (ya que actúan como un tampón químico), Transducción de señales (Ej: rodopsina) Protectora o defensiva (Ej: trombina y fibrinógeno) Las proteínas están formadas por aminoácidos los cuales a su vez están formados por enlaces peptídicos para formar esfingosinas. Las proteínas de todos los seres vivos están determinadas mayoritariamente por su genética (con excepción de algunos péptidos antimicrobianos de síntesis no ribosomal), es decir, la información genética determina en gran medida qué proteínas tiene una célula, un tejido y un organismo. Las proteínas se sintetizan dependiendo de cómo se encuentren regulados los genes que las codifican. Por lo tanto, son susceptibles a señales o factores externos. El conjunto de las proteínas expresadas en una circunstancia determinada es denominado proteoma.
Este método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde. Al final del proceso se obtendrán 2 moléculas de ADN, cada una de ellas formada por una cadena original y una cadena complementaria que ha sido sintetizada
PCR El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Desnaturalización Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación, Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada
ACIDOS NUCLEICOS
Los ácidos nucleicos son grandes polímeros formados por la repetición de monómeros denominados nucleótidos, unidos mediante enlaces fosfodiéster. Se forman, así, largas cadenas; algunas moléculas de ácidos nucleicos llegan a alcanzar tamaños gigantescos, con millones de nucleótidos encadenados. Los ácidos nucleicos almacenan la información genética de los organismos vivos y son los responsables de la transmisión hereditaria. Existen dos tipos básicos, el ADN y el ARN.
El ADN MITOCONDRIAL
Es un fragmento de ácido nucleico encontrado dentro de las mitocondrias. Las mitocondrias son pequeños organelos o estructuras celulares que se encuentran dentro de las células eucarióticas (generalmente las células animales). Estos organelos se usan básicamente para la producción y generación de energía para que la célula lleve a cabo sus funciones. Las plantas tienen cloroplastos. Las células animales tienen mitocondrias. Y cada una de estas mitocondrias tiene su propio genoma. Su propia molécula circular de ADN. Y eso es el ADN mitocondrial, porque en realidad es diferente del ADN que se encuentra en el núcleo de las células. No se encuentra dentro del núcleo. El número de genes en el ADN mitocondrial es de 37, frente a los 20.000 - 25.000 genes del ADN cromosómico nuclear humanos.Codifica dos ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa. El ADN mitocondrial está en replicación constante, independientemente del ciclo y del tipo celular.
ELISA DIRECTA E INDIRECTA Un ensayo de inmunoabsorción ligado a enzimas (ELISA) implica la reacción de un complejo anticuerpo-enzima con un antígeno o anticuerpo que se mantiene inmóvil sobre una superficie sólida. En la incubación de la enzima conjugada con un sustrato adecuado, un producto es formado. La formación de un producto ayuda a detectar la presencia del antígeno o anticuerpo y su cantidad proporciona una medida de la cantidad de antígeno-anticuerpo de la reacción que se produjo. La prueba de ELISA se puede realizar de dos formas: directa e indirecta.
ADN Mitocondrial:También llamado cromosoma mitocondrial, es una molécula circular de DNA de un tamaño de 16569 pares de bases (bp) (8000 veces menor que el cromosoma medio). Este tamaño de 16569 bp corresponde al primer ADN secuenciado (secuencia Cambridge) aunque existen otras variantes con un número de pares de bases que oscila entre 16559 y 16570. En cada mitocondria existen varias copias de este ADN, de modo que el número de cromosomas mitocondriales en cada célula puede ser de varios miles. Cuatro o cinco cromosomas mitocondriales se agrupan formando los llamados nucleoides. El ADN mitocondrial es muy parecido al del los cromosomas bacterianos, estando formado por dos cadenas complementarias, cada una con 16569 pares de bases, pero con un peso molecular diferente: la cadena pesada H (peso molecular, 5.168.726 daltons) contiene muchas más G que la cadena ligera L (peso molecular, 5.060.609 daltons). La mayor parte de la cadena H constituye el molde para la transcripción de la mayor parte de los genes, mientras que la cadena L es la cadena codificadora.El genoma mitocondrial contiene un total de 37 genes de los cuales 13 genes que codifican para ARNs mensajeros, y por lo tanto para 13 proteínas, 22 genes que codifican para 22 tARNs (ARNs de transferencia, que se representan simbólicamente como hojas de trebol) y 2 genes que codifican para dos rRNAs mitocondriales (RNAs ribosómicos). En la práctica, cuando se secuencia el ADN miocrondrial se unna persona en particular, se observan un cierto número de variaciones que son simplemente polimorfismos no tienen ninguna consecuencia clínica. De hecho, una región del ADN-mitocondrial llamada región de control, es tan polimórfica que se utiliza en medicina forense para identificar al sujeto.Además de las proteínas que la mitocondria puede sintetizar por si misma, necesita importar algunas otras sintetizadas en el núcleo. De igual forma, los lípidos que forman las membranas externa e interna de la mitocondria son importadas.Las mutaciones de algunos de los genes mitocondriales ocasionan enfermedades en el hombre. Se conocen las siguientes mutaciones:MELAS: (miopatía mitocondrial con encefalopatía, acidosis láctica y episodios similares al ictus). Se debe a una disfunción el complejo I de la cadena respiratoria mitocondrial debida a un cambio de bases en el par 3243 de la cadena pesadaMERRF: (epilepsia mioclónica, fibras rojas deshilachadas): se debe sobre todo a una mutación del gen que codifica el t-ARN de la lisina por un cambio de bases en la posición 8344 de la cadena pesada. Este cambio produce una disfunción del complejo V de la cadena respiratoria.
ARN El ácido ribonucleico (ARN o RNA) es un ácido nucleico formado por una cadena de ribonucleótidos. Está presente tanto en las células procariotas como en las eucariotas, y es el único material genético de ciertos virus (virus ARN). El ARN celular es lineal y de hebra sencilla, pero en el genoma de algunos virus es de doble hebra. En los organismos celulares desempeña diversas funciones. Es la molécula que dirige las etapas intermedias de la síntesis proteica; el ADN no puede actuar solo, y se vale del ARN para transferir esta información vital durante la síntesis de proteínas (producción de las proteínas que necesita la célula para sus actividades y su desarrollo). Varios tipos de ARN regulan la expresión génica, mientras que otros tienen actividad catalítica. El ARN es, pues, mucho más versátil que el ADN.
ADN El ácido desoxirribonucleico, abreviado como ADN, es un ácido nucleico que contiene instrucciones genéticas usadas en el desarrollo y funcionamiento de todos los organismos vivos conocidos y algunos virus, y es responsable de su transmisión hereditaria. El papel principal de la molécula de ADN es el almacenamiento a largo plazo de información. Muchas veces, el ADN es comparado con un plano o una receta, o un código, ya que contiene las instrucciones necesarias para construir otros componentes de las células, como las proteínas y las moléculas de ARN. Los segmentos de ADN que llevan esta información genética son llamados genes, pero las otras secuencias de ADN tienen propósitos estructurales o toman parte en la regulación del uso de esta información genética.
Se denomina compuesto químico inorgánico a todos aquellos compuestos que están formados por distintos elementos, pero en los que su componente principal no siempre es el carbono, siendo el agua el más abundante. En los compuestos inorgánicos se podría decir que participan casi la totalidad de elementos conocidos. como el oro,plata . Mientras que un compuesto orgánico se forma de manera natural tanto en animales como en vegetales, uno inorgánico se forma de manera ordinaria por la acción de varios fenómenos físicos y químicos: electrólisis, fusión, etc. También podrían considerarse agentes de la creación de estas sustancias a la energía solar, el agua, el oxígeno. Los enlaces que forman los compuestos inorgánicos suelen ser iónicos o covalentes. Ejemplos de compuestos inorgánicos: Cada molécula de cloruro de sodio (NaCl) está compuesta por un átomo de sodio y otro de cloro. Cada molécula de agua (H2O) está compuesta por dos átomos de hidrógeno y uno de oxígeno. Cada molécula de amoníaco (NH3) está compuesta por un átomo de nitrógeno y tres de hidrógeno.
El primer paso de todo Western Blot es la separación de las macromoléculas mediante geles de electroforesis; después de la misma, las macromoléculas ya separadas en función de su diferente peso molecular se transfieren a una segunda matriz, generalmente una membrana de nitrocelulosa o PVDF. Posteriormente, se bloquea la membrana para evitar la unión inespecífica a su superficie de los anticuerpos que se van a utilizar para la detección de la proteína de interés. En el siguiente paso, se une a dicha proteína transferida un anticuerpo especifico marcado con una enzima y, finalmente, se añade un sustrato apropiado para dicha enzima con lo que se produce un producto detectable como, por ejemplo, un precipitado cromogénico o fluorogénico en la membrana. En la actualidad, los métodos más sensibles emplean sustratos quimioluminiscentes que cuando se combinan con la enzima correspondiente, producen luz como producto final, que puede detectarse mediante una película o una cámara CCD. En todos los casos y sea cual sea el sustrato que se utilice, la intensidad de la señal se correlaciona con la cantidad del antígeno en la superficie de la membrana.
TEMA10: ELECTROFORESIS DE PROTEÍNAS EN SUERO Este examen mide los tipos de proteína en la parte líquida (suero) de una muestra de sangre. Ver también: Forma en que se realiza el examen Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción
La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína. La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL).
Tema 3 - ELISA… El ensayo inmunoabsorbente ligado a enzimas (ELISA) utiliza como sus siglas lo indican una enzima como marcador para mediar la formación de complejos antígeno-anticuerpo. Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario. Casi todas las pruebas ELISA son ensayos en fase sólida en los cuales se adsorbe un antígeno o un anticuerpo sobre un soporte sólido. Algunos de los protocolos se basan en reacciones de enlace competitivo y otras en reacciones de enlace no competitivo, pero en todas las pruebas ELISA se requiere de un paso de separación para eliminar el conjugado enzimático libre antes de proceder a determinar la cantidad de conjugado enzimático enlazado. Para lo cual se añade sustrato enzimático y se mide la reacción catalítica entre la enzima y el sustrato. Por sus características catalíticas las enzimas son marcadores muy sensibles y versátiles. Una sola proteína enzimática puede transformar en algunos minutos gran número de moléculas de sustrato en una cantidad igualmente abundante de producto final, produciendo un cambio de color amplificado y que se detecta con facilidad.En el método ELISA de inhibición el cambio de color se reduce, porque la actividad enzimática se inhibe cuando el anticuerpo se enlaza al conjugado enzimático.
TEMA 7 :El Western blot, o inmunoblot, es una técnica analítica usada para detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis en gel se separan las proteínas atendiendo al criterio que se desee: peso molecular, estructura, hidrofobicidad, etc. Hay casi tantas posibilidades como tipos de electroforesis existen. Luego son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella.1 2 Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas. Esta técnica es hoy en día imprescindible en varios campos de la biología, como la biología molecular, la bioquímica, la biotecnología o la inmunología. Existe toda una industria especializada en la venta de anticuerpos (monoclonales y policlonales) contra decenas de miles de proteínas diferentes.3 4
MANEJO DE MUESTRAS Y REACTIVOS. El laboratorio de biología celular y molecular tiene una serie de condiciones especiales relacionadas con el manejo de reactivos, elementos y equipos que pueden influir en la salud del personal así como repercutir en el medio ambiente. Por ende, es importante conocer las normas establecidas, familiarizarse con el uso de cada uno de los materiales de trabajo para evitar cometer errores o sufrir accidentes durante su manipulación.
El personal del laboratorio, por lo tanto, está expuesto a una serie de riesgos ocupacionales por agentes físicos, químicos y biológicos, los cuales pueden originar alteraciones en la salud, generalmente prevenibles si se aplican las normas de bioseguridad, que incluyen el manejo correcto de material infeccioso, así como el uso de elementos protectores, y el manejo adecuado de desechos biológicos y reactivos químicos. Al manipular reactivos químicos, es necesario conocer y evaluar sus peligros y determinar la forma apropiada para su manejo seguro. Esta información se obtiene de las etiquetas, hojas de seguridad suministradas por el fabricante u otras fuentes. Las etiquetas del fabricante incluyen el nombre del producto, el pictograma de seguridad, las frases de riesgo entre otras. El etiquetado del puesto de trabajo se emplea cuando el producto es transferido a otro contenedor diferente al original. Allí debe contener el nombre del producto, del fabricante, frases de riesgo y consejos de prudencia.
Las hojas de seguridad informan sobre las propiedades físicas, químicas y toxicológicas de una sustancia y los procedimientos recomendados para una manipulación segura, uso de equipos para protección, primeros auxilios, tratamiento de desechos, derrames y otros.
La clasificación de las sustancias químicas depende de su peligrosidad, de lo cual se establece que existen:
• Tóxicas • Muy tóxicas • Corrosivas • Irritantes • Inflamables • Extremadamente inflamables Los riegos biológicos se pueden identificar según: el tipo de material biológico que se manipula, los procedimientos de rutina realizados en el laboratorio, el manejo de desechos biológicos, el aseo y desinfección de las áreas de trabajo, la disponibilidad de implementos de protección personal, los protocolos de reconocimiento, las enfermedades relacionadas con el trabajo, los accidentes e incidentes más comunes como pinchazos, cortaduras e inhalaciones, las enfermedades ocupacionales adquiridas en el laboratorio.
REACCION EN CADENA DE LA POLIMERASA. Durante las décadas de 1970-1980, la manera más práctica de hacer múltiples copias de una secuencia particular de ADN era introduciendo una molécula de ADN recombinante (un plásmido más un gen) en una célula huésped. Esto podía resultar un poco engorroso, ya que muchas veces se disponía de una cantidad muy pequeña de moléculas de ADN para realizar pruebas.
A mediados de la década de los 80, la invención de una técnica capaz de generar centenares de miles de copias de una secuencia sin la necesidad de clonarla en ningún tipo de vector resolvió este problema. Kary Mullis, un bioquímico americano que trabajaba sintetizando ADN, resolvió este problema con la invención de la reacción en cadena de la polimerasa.
Esta se basa en la separación de ambas hebras de ADN, la posterior unión de pequeños fragmentos (denominados primers) y la extensión de estos fragmentos usando de molde el ADN de la muestra. Así se obtienen dos copias idénticas por cada molécula en cada ciclo de reacción. Para llevar a cabo esta reacción se requiere de una enzima polimerasa que sea capaz de soportar la alta temperatura del proceso de desnaturalización. Esta enzima es una ADN-polimerasa especial obtenida de una bacteria termófila (la Thermus aquaticus), resistente a altas temperaturas.
La reacción en cadena de la polimerasa permite amplificar la muestra de manera espectacular y su sensibilidad es tal que alcanza con una molécula para que la reacción genere una infinidad de copias. Esto la convierte en una importante, si no imprescindible, herramienta para el análisis de filiación o de criminalística forense, ya que con sólo una pequeñísima muestra se pueden realizar diferentes estudios comparativos que nos permiten conocer el dueño de un “rastro” genético en particular.
También se usa para el desarrollo de nuevas estrategias de diagnóstico médico, como la detección de virus o de mutaciones que provocan enfermedades genéticas, a partir de una muestra de ADN tan pequeña como un cabello o una gota de sangre.
Otra aplicación de la técnica de la reacción en cadena de la polimerasa es la transcripción inversa de ARNm. Mediante el uso de una enzima de origen viral denominada transcriptasa reversa puede usarse como molde una molécula de ARNm, transcribirla a una secuencia de ADN, y luego ser amplificada como cualquier otra secuencia por la reacción en cadena de la polimerasa, con lo que se obtiene una gran cantidad de ADN a partir de unas pocas moléculas de ARNm. Esta técnica es muy empleada para el estudio de expresión génica, así como para la producción de proteínas en diferentes organismos.
ETAPAS DE LA PCR Desnaturalización: Para que pueda iniciarse la reacción es preciso que las moléculas de ADN molde se encuentren en forma de cadena simple. Esto se consigue calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrogeno intercatenarios y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación.
Hibridación: Una vez que el ADN está desnaturalizado se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
Extensión: Durante esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la “Taq polimerasa” alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
Las enzimas de restricción son nucleasas que cortan ADN de doble cadena cuando reconocen un patrón de secuencia específico. Generan fragmentos de ADN conocidos como fragmentos de restricción. Las enzimas de restricción son herramientas imprescindibles en biología molecular, ingeniería genética y biotecnología.
Las enzimas de restricción son nucleasas que cortan el ADN cuando reconocen un patrón de secuencia específico.
Se descubrieron en los años 60 y su uso en biología molecular empezó en los 70 permitiendo el desarrollo de las tecnologías de ADN recombinante. Las enzimas de restricción tienen actividad endonucleasa y cortan enlaces fosfodiéster en las dos cadenas del ADN. Reconocen ciertas secuencias en el ADN. Las enzimas de restricción de tipo I y III reconocen una secuencia específica pero cortan a una distancia variable del sitio de reconocimiento (sitio de restricción). Las enzimas de restricción de tipo II reconocen una secuencia específica conocida como secuencia diana y siempre cortan entre los mismos nucleótidos. De ahí que generen siempre los mismos fragmentos de restricción. La secuencia diana es de tamaño variable, la mayoría de 4 y 6 nucleótidos, y con frecuencia es parcialmente palindrómica. Algunas enzimas de restricción cortan generando extremos llamados romos mientras que otras cortan generando extremos llamados cohesivos.
Las enzimas de restricción son una herramienta básica en Biología molecular: en laboratorios de PCR, creación de sondas para hibridación, laboratorios de secuenciación de ADN, ingeniería genética, procesos de clonación, manejo de genotecas y en muchas otras áreas de genómica.
BRENDA Las enzimas de restricción, también conocidas como endonucleasas, son enzimas que cortan los enlaces fosfodiester del material genético a partir de una secuencia que reconocen.
· Las mismas permiten cortar DNA de hebra doble, donde reconocen secuencias palindrómicas (secuencias que se leen igual en ambas direcciones).
· Son extraídas de organismos procarióticos (bacterias), donde actúan como un mecanismo de defensa, para degradar material genético extraño que entre en la célula. Las bacterias tienen la capacidad de metilar su DNA, lo cual sirve para distinguir entre el DNA extraño y el DNA propio. Las enzimas de restricción no pueden cortar DNA metilado, de este modo solo afectan el DNA extranjero y no el DNA bacterial. EcoRI es una enzima de restricción producida por el microorganismo Escherichia coli que posee una diana de restricción en el ADN de cadena doble dependiente de una secuencia metilada (al estar la hebra metilada, no hay corte), palindrómica y asimétrica, sobre la cual su actividad catalítica hidrolasa genera extremos cohesivos. Es sin duda la enzima más conocida dentro del mundo académico. La forma correcta de nombrarla es "Eco erre 1". En adision es la mas utilizada en laboratorios de universidades para beneficiar al estudiante y que aprenda como manejar con enzimas de restricción.
ELISA directo (ensayo ELISA simple de dos capas). Las placas ELISA se preparan recubriendo los pocillos con las soluciones en las que se sospecha se encuentra el antígeno. Se incuban con anticuerpos marcados. Indican la presencia de antígeno en la solución analizada. Es necesario incluir controles negativos que serán muestras del mismo tipo de las analizadas (sangre, orina,...) pero en las que se tenga la certeza de la ausencia del antígeno buscado. Asimismo se incluyen controles positivos (soluciones donde se encuentra el antígeno buscado).
ELISA ''sándwich'' (Ensayo de captura de antígeno y detección mediante inmunocomplejos). Se trata de un ensayo muy empleado en el que se recubre el pocillo con un primer anticuerpo anti-antígeno. Después de lavar el exceso de anticuerpo se aplica la muestra problema en la que se encuentra el antígeno, que será retenido en el pocillo al ser reconocido por el primer anticuerpo. Después de un segundo lavado que elimina el material no retenido se aplica una solución con un segundo anticuerpo anti-antígeno marcado. Así pues cada molécula de antígeno estará unida a un anticuerpo en la base que lo retiene y un segundo anticuerpo, al menos, que lo marca. Este ensayo tiene una gran especificidad y sensibilidad debido a la amplificación de señal que permite el segundo anticuerpo.
ELISA indirecto. Las placas ELISA se preparan de la misma forma a la anterior. Los controles positivos y negativos son los mismos. El sistema de detección emplea dos anticuerpos: uno primario contra el antígeno y uno secundario marcado contra el primario. La detección tiene mayor sensibilidad por presentar una amplificación de señal debida a la unión de dos o más anticuerpos secundarios por cada primario. Es el ensayo más popular, como lo es la inmunofluorescencia indirecta, pues un mismo secundario marcado y un mismo sistema enzimático permite cuantificar una gran variedad de antígenos, por eso es un método más polivalente y barato, aunque se pierda algo de precisión por tener un eslabón más con respecto al método directo. La dilución de la solución que contiene el anticuerpo primario (por ejemplo: suero sanguíneo) es un factor muy importante a tener en cuenta para evitar la aparición de falsos negativos, ya que si la muestra está muy diluida no saldrá positiva si la titulación de anticuerpos es muy baja. Es decir, aunque los anticuerpos están presentes, la prueba no da positivo porque la concentración de anticuerpos específicos contra el antígeno que está pegado en el fondo del pocillo no es suficiente como para dar una señal detectable. El ELISA indirecto es el método de elección para detectar la presencia de anticuerpos séricos contra el virus de la inmunodeficiencia humana (VIH), agente causante del síndrome de inmunodeficiencia adquirida (SIDA). Según esta técnica, proteínas recombinantes de la envoltura y el núcleo del VIH se absorben como antígenos en fase sólida a los pocillos. Las personas afectadas de VIH producen anticuerpos séricos contra epítopos en estas proteínas víricas. En general, el ELISA indirecto permite detectar anticuepos séricos contra VIH desde las primeras seis semanas de una infección.
ALESHA LOW 2012-1318 ELISA INDIRECTO En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA.
SOUTHERN BLOT PARA DNA Southern blot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda. Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern.1. Extracción del ADN Se puede extraer ADN de casi cualquier tejido humano. Las posibles fuentes de ADN en la escena de un delito incluyen sangre, semen, tejido de una víctima muerta, células del folículo capilar y saliva. El ADN extraído de las pruebas del delito ("indicios" o "vestigios" biológicos) se compara con el extraído de muestras de referencia, obtenidas de personas conocidas,habitualmente de la sangre. 2. Digestión del ADN con una endonucleasa de restricción El ADN extraído de la muestra se trata con una endonucleasa de restricción,que es una enzima que corta el ADN bicatenario en donde tenga una secuencia característica. La enzima que se usa más frecuentemente para el análisis legal es HaeIII, que corta el ADN en la secuencia 5'-GGCC-3'. 3. Electroforesis en gel de agarosa Tras la digestión del ADN, los fragmentos de ADN resultantes se separan según su tamaño mediante electroforesis en geles de agarosa. Durante la electroforesis, las moléculas de ADN, que poseen carga negativa, migran hacia el electrodo positivo. Al avanzar las moléculas de ADN, su velocidad de migración se ve reducida por la matriz del gel de agarosa. Las moléculas menores se mueven más deprisa a través de los poros del gel que las de mayor tamaño. Como resultado, se produce una separación continua de los fragmentos de ADN de acuerdo con su tamaño, de modo que los fragmentos más pequeños avanzan la mayor distancia con referencia al origen o punto de aplicación de la muestra. 4. Preparación de un ensayo de Southern ("Southern blot") Tras la electroforesis, las moléculas de ADN separadas se desnaturalizan mientras permanecen en el gel de agarosa, impregnando éste con una disolución alcalina. Tras la neutralización de ésta, el DNA monocatenario resultante se transfiere a la superficie de una membrana de nailon,realizando así una copia o "calco" (la traducción más literal del inglés blot,conservando este sentido, es "secante"). Este proceso de desnaturalización y transferencia se conoce como método de Southern en recuerdo de quien lo inventó, Edward Southern. Al igual que la aplicación de un secante a un papel con la tinta húmeda transfiere una réplica de la imagen del papel al secante,el "calco" del ADN en el gel a la membrana de nailon conserva la distribución espacial de los fragmentos de ADN conseguida en el gel como resultado de la electroforesis.
Northern blot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente. El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southern blot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northern blot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.El procedimiento general comienza con la extracción del RNA total de una muestra de tejido homogenizado. El mRNA se puede aislar a través del uso de cromatografía para mantener solamente los ARN con colas de poli-A. Las muestras de ARN son entonces separadas por electroforesis en gel. Dada la fragilidad de los geles y la incapacidad de las sondas para penetrar en la matriz, las muestras de RNA, separadas por tamaño tras la electroforesis, serán tranferidas a una membrana de Nailon por medio de capilaridad o un sistema de transferencia al vacío.
Los sistemas de capilaridad iónica se utilizan pra transferir el ARN desde un gel de electroforesis a una membrana de Northern blot. Una membrana de Nylon cargada positivamente es el soporte mas efectivo para usar en el northern blot ya que los ácidos nucleicos, que están cargados negativamente, tienen una alta afinidad por estas membranas. El buffer de transferencia usado para el ensayo suele contener formamida, dado que ésta consigue disminuir la temperatura de hibridación de la sonda, lo cual previene la degradación del ARN por las altas temperaturas. Una vez que el ARN ha sido transferido a la membrana, este es inmovilizado a través de enlaces covalentes formados con la membrana, lo cual se consigue por medio de luz ultravioleta o calor. Después del marcaje de la sonda, ésta se hibrida con el RNA en la membrana. Las condiciones experimentales que pueden afectar la eficiencia y especificidad de la hibridación están determinadas por las condiciones iónicas, de viscosidad, la presencia de dobles hebras de RNA, bases desemparejadas y composición de las mismas. Finalmente, la membrana debe de lavarse para asegurar que la sonda se ha unido de forma específica y para evitar ruido de fondo en las señales emitidas por la misma. Estas señales serán detectadas por Rayos X y pueden ser cuantificadas mediante técnicas de densitometría. Para crear controles y poder asegurarnos de que no se están mostrando genes que no nos interesen se puede realizar posteriormente la determinación por microarrays o RT-PCR.
EASTERN BLOT es una técnica bioquímica utilizado para analizar proteínas modificaciones post traduccionales (PTM), tales como lípidos, phosphomoieties y glicoconjugados. Con mayor frecuencia se utiliza para detectar carbohidratos epítopos . Por lo tanto, este secante puede ser considerado una extensión de la técnica de bioquímica de transferencia Western . Varias técnicas han sido descritas por el término del Este secante, proteínas más uso borrados de SDS-PAGE en gel en un PVDF o nitrocelulosa membrana. Proteínas transferidas se analizaron para las modificaciones post-traduccionales utilizando sondas que pueden detectar los lípidos , hidratos de carbono , fosforilación o cualquier otra modificación de proteínas. Este Blot se debe usar para referirse a métodos que detectan sus objetivos a través de la interacción específica de la PTM y la sonda, distinguiéndolos de un estándar de transferencia Far-Western . En principio, este secante es similar a lectina secante (es decir, la detección de epítopes de carbohidratos en las proteínas o lípidos).
El Western blot, o inmunoblot, es una técnica analítica usada para detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis en gel se separan las proteínas atendiendo al criterio que se desee: peso molecular, estructura, hidrofobicidad, etc. Hay casi tantas posibilidades como tipos de electroforesis existen. Luego son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella.Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas. Esta técnica es hoy en día imprescindible en varios campos de la biología, como la biología molecular, la bioquímica, la biotecnología o la inmunología. Existe toda una industria especializada en la venta de anticuerpos (monoclonales y policlonales) contra decenas de miles de proteínas diferentes. El Western blot fue desarrollado en el laboratorio de George Stark, en la Universidad de Stanford. El nombre (Western, occidental en inglés) le fue dado por W. Neal Burnette, y consiste en un juego de palabras con una técnica análoga pero que usa DNA, el Southern (sureño) blot, que en este caso debe su nombre a su descubridor, Edwin Southern. Otras técnicas que fueron nombradas siguiendo este criterio son el Northern blot (en el que se separa e identifica RNA), el Eastern blot, el Southwestern blot.
El Southwestern blot es una técnica de laboratorio que permite la identificación y caracterización de proteínas por su capacidad de unión a fragmentos específicos de ADN. Para ello, se transfiere el resultado de una electroforesis a una membrana y se añaden oligonucleótidos para observar la unión a las proteínas. Fue descrito por primera vez en 1981 en el laboratorio de Brian Bowen. Su nombre se debe a la convergencia entre el Southern blot, que emplea sondas de ADN para detectar, en su caso, ADN fijado en una membrana, y el Western blot, en el cual se detectan proteínas fijadas a una membrana, aunque en este caso se realice con anticuerpos.
EXTRACCION DE DNA MITOCONDRIAL También llamado cromosoma mitocondrial, es una molécula circular de DNA (*) de un tamaño de 16569 pares de bases (bp) (8000 veces menor que el cromosoma medio). Este tamaño de 16569 bp corresponde al primer ADN secuenciado (secuencia Cambridge) aunque existen otras variantes con un número de pares de bases que oscila entre 16559 y 16570. En cada mitocondria existen varias copias de este ADN, de modo que el número de cromosomas mitocondriales en cada célula puede ser de varios miles. Cuatro o cinco cromosomas mitocondriales se agrupan formando los llamados nucleoides
El ADN mitocondrial es muy parecido al del los cromosomas bacterianos, estando formado por dos cadenas complementarias, cada una con 16569 pares de bases, pero con un peso molecular diferente: la cadena pesada H (peso molecular, 5.168.726 daltons) contiene muchas más G que la cadena ligera L (peso molecular, 5.060.609 daltons). La mayor parte de la cadena H constituye el molde para la transcripción de la mayor parte de los genes, mientras que la cadena L es la cadena codificadora (*)
El genoma mitocondrial contiene un total de 37 genes de los cuales 13 genes que codifican para ARNs mensajeros, y por lo tanto para 13 proteínas, 22 genes que codifican para 22 tARNs (ARNs de transferencia, que se representan simbólicamente como hojas de trebol) (*) y 2 genes que codifican para dos rRNAs mitocondriales (RNAs ribosómicos). En la práctica, cuando se secuencia el ADN miocrondrial se unna persona en particular, se observan un cierto número de variaciones que son simplemente polimorfismos no tienen ninguna consecuencia clínica. De hecho, una región del ADN-mitocondrial llamada región de control, es tan polimórfica que se utiliza en medicina forense para identificar al sujeto.
Además de las proteínas que la mitocondria puede sintetizar por si misma, necesita importar algunas otras sintetizadas en el núcleo. De igual forma, los lípidos que forman las membranas externa e interna de la mitocondria son importadas.
Las mutaciones de algunos de los genes mitocondriales ocasionan enfermedades en el hombre. Se conocen las siguientes mutaciones:
MELAS: (miopatía mitocondrial con encefalopatía, acidosis láctica y episodios similares al ictus). Se debe a una disfunción el complejo I de la cadena respiratoria mitocondrial debida a un cambio de bases en el par 3243 de la cadena pesada MERRF: (epilepsia mioclónica, fibras rojas deshilachadas): se debe sobre todo a una mutación del gen que codifica el t-ARN de la lisina por un cambio de bases en la posición 8344 de la cadena pesada. Este cambio produce una disfunción del complejo V de la cadena respiratoria NARP (neuropatía, ataxia, retinitis pigmentaria): se debe a una mutación del gen que codifica el complejo V de la cadena respiratoria (ATP-asa 6) LHON (neuropatía hereditaria de Leber): se debe a multiples mutaciones en los genes que codifican el complejo I (NADH-deshidrogenasa) Adicionalmente se han encontrado estas y otras mutaciones de los genes mitocondriales en muchos otros desórdenes (p. ejemplo, sordera, síndrome de Ham, etc).
La electroforesis se utiliza para identificar la presencia de proteínas anómalas, para identificar si falta alguna de las proteínas normales y para determinar si diferentes grupos de proteínas en sangre están aumentados o disminuidos.
Alteraciones del patrón obtenido característicamente permiten orientar el diagnóstico hacia una enfermedad u otra. La presencia de una anomalía en un patrón electroforético raramente es diagnóstica por sí misma, aunque sí proporciona una pista. El estudio en estos casos se ampliará con la finalidad de identificar la naturaleza de la enfermedad subyacente.Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina). Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (-) o cátodo (+) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración. Isoelectroenfoque: en lugar de separar las proteínas en función de su carga a un pH dado, se separan en función de su punto isoeléctrico (pI): el pI es el pH en el que la carga neta de la proteína es nula, y depende de la composición aminoacídica de la proteína. Se crea un gradiente de pH mediante anfolitos (estabilizan el pH a lo largo del gel). Cada proteína migrará hasta alcanzar su pI, punto en el cual precipitará al acumularse (de ahí el nombre, isoelectroenfoque). También se suelen utilizar acoplados a zimogramas si deseamos determinar el pI de una enzima o en electroforesis bidimensional como primera dimension. Separación por tamaño: permite separar proteínas y ácidos nucleicos. En el caso de las proteínas, deben ser tratadas con SDS (sodio dodecil sulfato) para que su carga sea negativa y todas migren hacia el ánodo (no es necesario hacer eso con los ácidos nucleicos, ya que tienen carga negativa); la separación se hace en medios de soporte en el que se ha creado un tamiz molecular (matriz), que hace que las proteínas más pequeñas corran más que las más grandes.
Tema 2 :Reacción de cadena polimerasa (PCR) La reacción en cadena de la polimerasa, ya más conocida como PCR, es una técnica que permite replicar entre cientos de miles y millones de veces, en el transcurrir de pocas horas e in vitro, pequeñas cantidades de ADN. Uno de los aportes fundamentales de esta metodología a la investigación básica en biología molecular consiste, precisamente, en la rapidez y eficiencia mediante las cuales se realiza una tarea que antes requería largas y tediosas jornadas. El producto quese obtiene al finalizar la reacción es una gran cantidad de un fragmento génico con alto grado de pureza- favorece la tarea de los investigadores empeñados en ampliar nuestros conocimientos sobre la estructura y función de los genes. El método se basa, en su forma más simple en la realización de tres reacciones sucesivas llevadas a cabo a distintas temperaturas. Estas reacciones se repiten cíclicamente entre veinte y cuarenta veces (véase la figura 1). La muestra se calienta, en el primer paso, hasta lograr la separación de las dos cadenas que constituyen el ADN, hecho que se conoce como "desnaturalización". En el segundo paso, la temperatura se reduce para permitir el "apareamiento" de cada una de dos cadenas cortas de nucleótidos (oligonucleóridos) con cada una de las hebras separadas del ADN molde. Se trata de segmentos de ADN de cadena simple, sintetizados en el laboratorio y diseñados de manera tal que permiten definir los límites del tramo de ADN que se desea replicar. Obviamente, para que se pueda producir el apareamiento, cada uno de estos oligonucleótidos, a los que se denomina "iniciadores" o primers, debe ser complementario del tramo al que tienen que unirse en las cadenas separadas del ADN molde. En tercer lugar, una enzima ADN polimerasa extiende los primers, en el espacio comprendido entre ambos, sintetizando las secuencias complementarias de las hebras del ADN molde. Para ello, la ADN polimerasa usa desoxidonucleósidos trifosfato agregados a la mezcla de reacción. La temperatura a la que se realiza el tercer paso está condicionada por aquélla a la cual "trabaja" la enzima ADN polimerasa. Al cabo del primer ciclo de tres reacciones (desnaturalización, apareamiento, extensión) el tramo de ADN elegido se ha duplicado y el doble de su cantidad original se encuentra disponible para ser nuevamente replicado en un segundo ciclo. El resultado de la aplicación de numerosos ciclos "en cadena" da lugar a la amplificación geométrica del segmento de ADN delimitado por los primers.Para replicar el ADN, la técnica PCR hizo uso, en un principio, de la ADN polimerasa de la bacteria Escherichia coli. Pero esta enzima resulta desactivada debido a la alta temperatura requerida para desnaturalizarla doble cadena del ADN, por lo cual debía agregarse enzima fresca al comenzar el tercer paso de cada ciclo. Este inconveniente fue solucionado de manera ingeniosa cuando se la reemplazó por su equivalente de la bacteria "termófila" Thermus aquaticus. En efecto, la ADN polimerasa de este microrganismo, denominada Taq polimerasa, actúa eficientemente entre los 75° C y los 80° C y resiste más de dos horas a 93° C. De esta manera es posible mezclar todos los componentes de la PCR al comenzar el primer ciclo y la reacción en cadena puede llevarse a cabo mediante equipos automatizados que realizarán los ciclos térmicos programados.
2012-1309 Tema 3: Enzimas de restrincion (BRENDA) Las enzimas de restricción son proteínas cuya función es cortar las hebras de ADN. Se podría decir que son “tijeras moleculares” que cortan ADN. Lo hacen en forma específica. Esto significa que cada enzima reconoce un sitio particular del ADN, es decir que reconoce una secuencia particular de nucleótidos. Esa secuencia específica para cada enzima se denomina “sitio de restricción”. Una vez que la enzima reconoce estos sitios, se posiciona sobre la molécula de ADN y corta dentro o en torno de esa secuencia. Las enzimas de restricción, conocidas también como endonucleasas, sólo cortan el ADN si reconocen en su interior una secuencia específica de nucleótidos. Estas enzimas fueron descubiertas en microorganismos. De hecho, se encuentran sólo en organismos procariotas (bacterias). Por esto, se les dio una nomenclatura asociada al organismo de donde provienen. Por ejemplo, las enzimas de restricción que se descubrieron en la bacteria Escherichia coli se denominan Eco. Existen diferentes tipos de enzimas Eco que se diferencian en la secuencia que reconocen y cortan. Para diferenciarlas se les agregan letras y números romanos, por ejemplo: Eco RI (Eco “erre” “uno”). Así, el sitio de restricción para EcoRI es la secuencia GAATTC, como muestra la ilustración anterior. Una vez que la enzima encontró ese sitio en el ADN, se acerca a la hebra de ADN y realiza el corte entre la G y la A. Al investigar otras especies de bacterias se descubrieron cientos de enzimas de restricción distintas y cada una reconoce una región específica. Estas enzimas fueron descubiertas en la década del ’70 y hasta la fecha existen más de 250 enzimas de restricción. Se cree que la función natural de estas enzimas en las bacterias es protegerlas contra ADN de virus que podrían ingresar en sus células. De esta manera, la bacteria utiliza estas “tijeras moleculares” para cortar en pedacitos el ADN viral que la infecta. El ADN propio de la bacteria no se corta, pues lo tiene “protegido” contra sus propias enzimas de restricción.
2012-1309 Tema 4: ELISA directo (Sandwish) La técnica de ELISA (Enzyme Linked InmunoSorbent Assay) es un procedimiento de ensayo inmuno enzimático. Se basa en la detección antígeno(Ag) o anticuerpo (Ac), La técnica de ELISA es un ensayo inmunoenzimático ampliamente empleado en el área médica para la cuantificación de moléculas especialmente de aquellas que experimentan cambios en diferentes estados como pueden ser infecciones por bacterias, virus, hongos o parásitos o fases activas de enfermedades autoinmunes. Como ejemplo se pueden medir por ELISA hormonas, autoanticuerpos, inmunoglobulinas contra antígenos de patógenos, toxinas, antígenos. Las técnicas de ELISA son también muy usadas en los ensayos de nuevos medicamentos para comprobar sus efectos cuantificando las moléculas de interés en cada caso. Existen numerosas variantes de este tipo de ensayo. Entre ellos se encuentran los métodos directo e indirecto. El método directo permite la detección del antígeno con un anticuerpo específico conjugado con una enzima como sistema de marcaje. Técnica: Las placas ELISA se preparan recubriendo los pocillos con las soluciones en las que se sospecha se encuentra el antígeno. Se incuban con anticuerpos marcados. Indican la presencia de antígeno en la solución analizada. Es necesario incluir controles negativos que serán muestras del mismo tipo de las analizadas (sangre, orina) pero en las que se tenga la certeza de la ausencia del antígeno buscado. Asimismo se incluyen controles positivos (soluciones donde se encuentra el antígeno buscado). Elisa sandwish Se trata de un ensayo muy empleado en el que se recubre el pocillo con un primer anticuerpo anti-antígeno. Después de lavar el exceso de anticuerpo se aplica la muestra problema en la que se encuentra el antígeno, que será retenido en el pocillo al ser reconocido por el primer anticuerpo. Después de un segundo lavado que elimina el material no retenido se aplica una solución con un segundo anticuerpo anti-antígeno marcado. Así pues cada molécula de antígeno estará unida a un anticuerpo en la base que lo retiene y un segundo anticuerpo, al menos, que lo marca. Este ensayo tiene una gran especificidad y sensibilidad debido a la amplificación de señal que permite el segundo anticuerpo.
2012-1309 Tema 5:ELISA Indirecto En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA. Es el ensayo más popular, como lo es la inmunofluorescencia indirecta, pues un mismo secundario marcado y un mismo sistema enzimático permite cuantificar una gran variedad de antígenos, por eso es un método más polivalente y barato, aunque se pierda algo de precisión por tener un eslabón más con respecto al método directo. La dilución de la solución que contiene el anticuerpo primario (por ejemplo: suero sanguíneo) es un factor muy importante a tener en cuenta para evitar la aparición de falsos negativos, ya que si la muestra está muy diluida nosaldrá positiva si la titulación de anticuerpos es muy baja. Es decir, aunque los anticuerpos están presentes, la prueba no da positivo porque la concentración de anticuerpos específicos contra el antígeno que está pegado en el fondo del pocillo no es suficiente como para dar una señal detectable.El ELISA indirecto es el método de elección para detectar la presencia de anticuerpos séricos contra el virus de la inmunodeficiencia humana (VIH), agente causante del síndrome de inmunodeficiencia adquirida (SIDA). Según esta técnica, proteínas recombinantes de la envoltura y el núcleo del VIH se absorben como antígenos en fase sólida a los pocillos. Las personas afectadas de VIH producen anticuerpos séricos contra epítopos en estas proteínas víricas. En general, el ELISA indirecto permite detectar anticuepos séricos contra VIH desde las primeras seis semanas de una infección.
2012-1309 Tema 6:Tecnicas Blotting El término “blotting” hace referencia a la transferencia de macromoléculas biológicas desde un gel hasta una membrana y su posterior detección en la superficie de la misma. Este proceso es necesario ya que todas estas macromoléculas están embebidas dentro de la matriz que forma el gel, lo que imposibilita realizar su detección, mientras que en la membrana, se encuentran accesibles al estar adheridas sobre la superficie de la misma. La técnica del Western Blot (también llamado “inmunoblotting” debido a que se utilizan anticuerpos para detectar el antígeno o los antígenos específicos de interés) fue descrita por primera vez por Towbin et al. en 1979 y en la actualidad es una técnica de rutina en todos los laboratorios que realizan análisis de proteínas. La especificidad de la unión antígeno – anticuerpo permite la detección de una única proteína dentro de una mezcla compleja de otras proteínas. En la actualidad se utiliza como criterio de identificación positivo de una proteína especifica en una mezcla compleja y para obtener datos cualitativos y semicuantitativos sobre la misma.
2012-1309 Tema 7:Electroforesis de proteínas Es un método de separación de proteínas mediante la aplicación de un campo eléctrico. Existen diferentes tipos en función del tipo de separación empleado: electroforesis de zona (separación en función de la carga), isoeléctrico enfoque y separación por tamaño en tamiz molecular (también aplicable a ácidos nucleicos). Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina).Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (-) o cátodo (+) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración.
2012-1309 Tema 8:El ADN mitocondrial Es un fragmento de ácido nucleico encontrado dentro de las mitocondrias. Las mitocondrias son pequeños organelos o estructuras celulares que se encuentran dentro de las células eucarióticas (generalmente las células animales). Estos organelos se usan básicamente para la producción y generación de energía para que la célula lleve a cabo sus funciones. Las plantas tienen cloroplastos. Las células animales tienen mitocondrias. Y cada una de estas mitocondrias tiene su propio genoma. Su propia molécula circular de ADN. Y eso es el ADN mitocondrial, porque en realidad es diferente del ADN que se encuentra en el núcleo de las células. No se encuentra dentro del núcleo. El número de genes en el ADN mitocondrial es de 37, frente a los 20.000 - 25.000 genes del ADN cromosómico nuclear humanos.Codifica dos ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa. El ADN mitocondrial está en replicación constante, independientemente del ciclo y del tipo celular. Se piensa que tiene lugar de forma asíncrona, es decir, que tiene lugar en las dos cadenas en tiempos diferentes y con dos orígenes distintos hacia direcciones contrarias. El comienzo tendría lugar en el origen de la cadena pesada, situado en el bucle D, y replicaría ésta tomando como molde la cadena ligera. Cuando se alcanza el segundo origen, situado a dos tercios de distancia del primero, comienza la segunda ronda de replicación en sentido opuesto. Se ha propuesto un nuevo sistema de replicación que coexistiría con el primero. Sería bidireccional y comportaría una coordinación entre hebras directas y retrasadas.
2012-1309 Tema 9: Anticuerpos monoclonales Son anticuerpos homogéneos producido por una célula híbrida producto de la fusión de un clon de linfocitos B descendiente de una sola y únicacélula madre y una célula plasmática tumoral. Los anticuerpos monoclonales mAB, de la frase en inglés con idéntico significado que en español: monoclonal antibody, son anticuerpos idénticos porque son producidos por un solo tipo de célula del sistema inmune, es decir, todos los clones proceden de una sola célula madre. Es posible producir anticuerpos monoclonales que se unan específicamente con cualquier molécula con carácter antigénico. Este fenómeno es de gran utilidad en bioquímica, biología molecular y medicina. Si una sustancia extraña (antígeno) se inyecta en el cuerpo de un ratón o un humano, alguna de las células B de su sistema inmune se transformarán en células plasmáticasy empezarán a producir anticuerpos que se unirán a ese antígeno. Cada célula B produce un solo tipo de anticuerpo, pero diferentes linfocitos B producirán anticuerpos estructuralmente diferentes que se unen a distintas partes del antígeno. Esta mezcla fisiológica natural de anticuerpos es conocida como Antisuero policlonal.Para producir anticuerpos monoclonales, primero se extraen células B del bazo de un animal que ha sido expuesto al antígeno. Estas células B son fusionadas en presencia de PEG (polietilenglicol) con células tumorales de mieloma múltiple (un tipo de cáncer) que pueden crecer indefinidamente en cultivo celular. Esta fusión hace a las membranas celulares más permeables. Estas células fusionadas híbridas, llamadas hibridomas pueden multiplicarse rápida e indefinidamente, puesto que son células tumorales después de todo y pueden producir gran cantidad de anticuerpos. Los hibridomas son suficientemente diluidos y cultivados para obtener un número diferente de determinadas colonias, las cuales producen sólo un tipo de anticuerpo. Los anticuerpos de diferentes colonias son analizados para conocer su capacidad de unirse a un antígeno determinado, por ejemplo con un tipo de test llamado ELISA, y para seleccionarse y aislarse de la manera más efectiva.











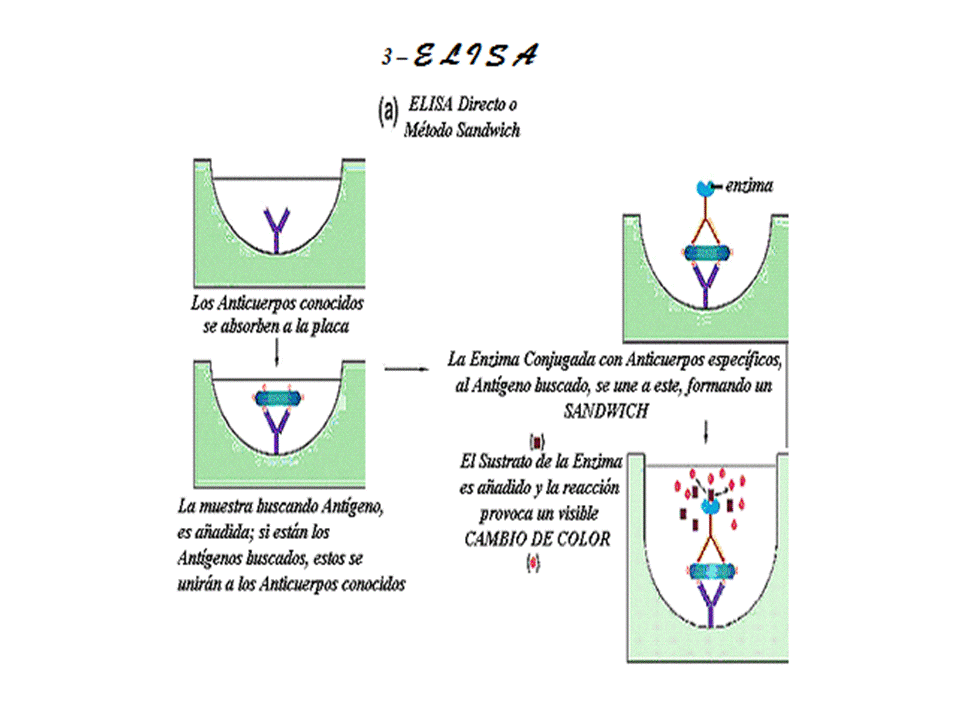













Este comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarRicardo Alayon 2012-1024
ResponderEliminar1)REACCIÓN EN CADENA DE LA POLIMERASA (PCR)
Este método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde. Al final del proceso se obtendrán 2 moléculas de ADN, cada una de ellas formada por una cadena original y una cadena complementaria que ha sido sintetizada. La enzima que realiza este proceso se denomina DNA polimerasa. Esta enzima añade los nucleótidos en el hidroxilo 3 terminal de la cadena de DNA paterna.
ETAPAS:
A)Desnaturalización: Se comienza calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrógeno y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación.
B)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
2- Enzima de restricción (BRENDA)
Es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
El mecanismo de corte de DNA se realiza a través de la ruptura de dos enlaces fosfodiéster en la doble hebra, lo que da lugar a dos extremos de DNA. Éstos pueden ser romos (cuando los enlaces rotos coinciden) o Cohesivos/escalonados. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
3 - ELISA…
ResponderEliminarEl ensayo inmunoabsorbente ligado a enzimas (ELISA) utiliza como sus siglas lo indican una enzima como marcador para mediar la formación de complejos antígeno-anticuerpo.
Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario. Casi todas las pruebas ELISA son ensayos en fase sólida en los cuales se adsorbe un antígeno o un anticuerpo sobre un soporte sólido. Algunos de los protocolos se basan en reacciones de enlace competitivo y otras en reacciones de enlace no competitivo, pero en todas las pruebas ELISA se requiere de un paso de separación para eliminar el conjugado enzimático libre antes de proceder a determinar la cantidad de conjugado enzimático enlazado. Para lo cual se añade sustrato enzimático y se mide la reacción catalítica entre la enzima y el sustrato. Por sus características catalíticas las enzimas son marcadores muy sensibles y versátiles. Una sola proteína enzimática puede transformar en algunos minutos gran número de moléculas de sustrato en una cantidad igualmente abundante de producto final, produciendo un cambio de color amplificado y que se detecta con facilidad.En el método ELISA de inhibición el cambio de color se reduce, porque la actividad enzimática se inhibe cuando el anticuerpo se enlaza al conjugado enzimático.
6-Eastern Blot
ResponderEliminarSe considera una extencion de la tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma.
4 - Southern Blot para ADN…
Southern blot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda. Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern. El sector de ADN buscando puede ser un gen, o parte del mismo. Se puede utilizar para el diagnostico molecular de patologias geneticas. LA doble hebra de ADN se forma por complementariedad SONDA mustra (una de las hebras es la sonda en simple caden y la otra es la muestra en simple cadena). Los nucleotidos de la sonda esta macados P 32 (radioactivos); si al revelar hay marcador radioactivo presente, esta en la muestra analizada la secuencia complementaria a la sonda. Pasos: extraccion de ADN. Fragmentar el ADN en porciones mas pequeñas (enzimas de resctriccion). Separar los ragmentos realizando una corrida electroforetica usando como sustrato de migracion un gel. (separacion por tamaño). Se agrega la sonda sobre la membrana y se incuba. Se lava para quitar el maximo de hibridaciones inespecificas. Se revela por autoradiografia.
5- Northern Blot para RNA
Northern blot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente.
El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southern blot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northern blot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.
7-Western blot para proteina
ResponderEliminarEl western blot es un método en biología molecular/bioquímica/inmunogenética para la detección de proteínas en una muestra de un tejido homogeneizado o extracto. Se utiliza una electroforesis para separar proteínas desnaturalizadas de la masa. Las proteínas son trasferidas desde el gel hacia la membrana (originalmente de nitrocelulosa), donde son examinadas utilizando anticuerpos específicos para la proteína. Como resultado, se pueden examinar la cantidad de proteínas en una muestra y comparar los niveles de presencia entre varios grupos. Otras técnicas ,también usando anticuerpos, permiten la detención de proteínas en tejidos (inmunohistoquímica) y células (inmunocitoquímica).
En el Northern blot, se utiliza para identificar las cadenas de ARN de secuencia semejante al ADN que se usa como sonda; a diferencia del tipo Southern que se utiliza para identificar ADN, el método northern sirve para identificar ARN.
De manera resumida, el Western sirve para identificar proteínas y el Northern para identificar secuencias de RNA.
Bueno, sus aplicaciones son muy variadas y un ejemplo que puedo decirte es el de la detección del VIH. De manera simple, el western blot se utiliza para detectar las proteínas de la membrana del virus, como dice en la descripción de arriba, estas proteínas provienen de la muestra (generalmente sangre que ya está infectada con el virus), las cuales son separadas por electroforesis en gel de poliacrilamida, y posteriormente detectadas por un anticuerpo específico (específico a la proteína, es decir, que sólo podrá unirse a esa proteína, a ninguna otra) en la membrana de nitrocelulosa.
Bueno, espero que te sirva de algo, lo primero lo saqué de wikipedia, pero me pareció muy escueto, por lo que decidí ampliarlo un poquito con lo que me acuerdo. Ah, si, y el northern detecta secuencias de RNA, y tiene aplicaciones similares, ya que como sabes, cuando hay síntesis de proteínas, existe un RNA mensajero que, junto con el RNA ribosomal y el de transferencia, que sintetizan a la proteína en el retículo endoplásmico... entonces también pueden detectarse un RNA mensajero específico para una proteína de interés, como en el caso del VIH...
8-Southern Blot para adn y proteina
ResponderEliminarEl Southern Blot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizando sonicación ó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAds ó RNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.El traspaso de las muestras desde el gel al filtro es una etapa esencial, porque el reconocimiento debases complementarias entre sonda y secuencias de DNA en el gel es muy ineficiente.Cabe señalar que el nombre de la técnica Southern Blot deriva en parte del apellido Southern delinvestigador que la desarrolló y de la palabra en inglés Blot que significa traspasar
9- Extracción de adn mitocondrial
ResponderEliminarEl ADN mitocondrial (mtDNA) es un material genético circular cerrado de doble cadena que se localiza en el interior de las mitocondrias celulares.El genoma mitocondrial consta de aproximadamente 16500 pares de bases (p.b.), y codifica para una pequeña fracción de las proteínas mitocondriales. El mtDNA contiene información de 38 genes: 2rRNA (12S y 16S), 22tRNA y 13 genes estructurales, los cuales codifican diferentes subunidades de los complejos enzimáticos del sistema de fosforilación oxidativa.La región mayor no codificante, conocida como región control o D-Loop, ocupa 1122 pares de bases. Esta región destaca por su elevada tasa de mutación y por ser muy variable entre las diferentes poblaciones. La variabilidad en la región control se concentra básicamente en tres regiones o segmentos hipervariables:
- HVSI (posiciones 16024-16365)
- HVSII (posiciones 73-340)
- HVSIII (posiciones 438-574)
De las tres, la más polimórfica es la HVSI, por lo que es la que se analiza principalmente en estudios de Antropología, Genética de Poblaciones y Medicina Forense.
El estudio del ADN mitocondrial resulta especialmente recomendado cuando se trabaja con muestras muy degradadas, como es de esperar en ADN antiguo. Se calcula que una célula puede contener hasta un centenar de mitocondrias, y que dentro de cada mitocondria coexisten entre 1000 y 10000 copias de ADN mitocondrial. Este elevado número de moléculas de ADN mitocondrial en la célula hace que su recuperación en aquellos casos en los que el ADN de partida es muy escaso o está muy degradado, sea mucho más eficiente que, por ejemplo, la de ADN nuclear o autosómico. En la especie humana, el sexo viene determinado por el tipo de cromosomas sexuales presentes en las células (cromosomas X e Y). Así, el sexo femenino viene definido por dos cromosomas X (XX) y el masculino por un cromosoma X y un cromosoma Y (XY).
A excepción de dos pequeñas regiones situadas en los extremos en las que existe cierto grado de recombinación con el cromosoma X, el cromosoma Y se transmite como una unidad íntegra de padres a hijos varones. Esta característica hace que, al igual que el ADN mitocondrial para las mujeres, pueda seguirse una determinada variante o haplotipo en sucesivas generaciones.
El Cromosoma Y se analiza de forma rutinaria en el campo de la Genética de Poblaciones y la Genética Forense. Sin embargo, su empleo en estudios de ADN antiguo es todavía muy restringido por motivos técnicos que tienen que ver con su escasez, dado que únicamente existe una copia de cromosoma Y por célula.
10- Electroresis de proteina
ResponderEliminarSe necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción
La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína.
La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL.
A usted le pueden solicitar no comer ni beber nada durante 12 horas antes de un examen de electroforesis de lipoproteínas.
El médico le puede solicitar que deje de tomar medicamentos que pudieran afectar el examen. NO suspenda ningún medicamento sin consultarlo previamente con éste.
Los medicamentos que pueden afectar la medición de las proteínas totales incluyen clorpromazina, corticoesteroides, isoniazida, neomicina, fenacemida, salicilatos, sulfamidas y tolbutamida.Cuando se inserta la aguja para extraer la sangre, algunas personas sienten un dolor moderado, mientras que otras sólo sienten un pinchazo o sensación de picadura. Posteriormente, puede haber algo de sensación pulsátil.Las proteínas están compuestas de aminoácidos y son componentes importantes de todas las células y los tejidos. Existen muchas clases de proteínas en el cuerpo, con muchas funciones diferentes. Los ejemplos de proteínas son: las enzimas, algunas hormonas, la hemoglobina, la lipoproteína de baja densidad (colesterol "malo") y otras.
Las proteínas séricas se clasifican como albúmina y globulinas. La albúmina es la proteína de mayor concentración en el suero. Trasporta muchas moléculas pequeñas, pero también es importante para impedir que el líquido se escape de los vasos sanguíneos hacia los tejidos.
Jhomairy Miller B. 2012-1419
ResponderEliminarLa Reacción en Cadena de la Polimerasa (PCR) se realiza en el laboratorio con el objetivo de obtener un gran número de copias de un fragmento de ADN a partir de una pequeña muestra de la original. También nos permite amplificar la cadena, de manera que sea más fácil identificar algún virus o enfermedad dentro de estas. Costa de tres fases: 1) Desnaturalización (se calienta la muestra para romper las cadenas), 2) Templado (bajar la temperatura para que los iniciadores se unan con otras cadenas) y 3) Polimerización (se eleva de nuevo la temperatura para copiar el acido nucleído).
Una enzima de restricción es laque puedes reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortarlo (atreves de la ruptura de dos enlaces fosfodiester en la doble hebra) en el sitio de restricción o no muy lejano a este. Estos sitios cuentan con 4 a 12 pares de bases. Los fragmentos que resultan del corte se pueden unir con otras enzimas llamada ligasas. La Enzima de Restricción o endonucleasa de restricción) se conoce como BRENDA en sus siglas en ingles Bacterial Restriction Endonuclease Analysis.
ELISA (del ingles Enzyme-Linked ImmunoSorbent Assay, Ensayo por inmunoabsorción ligado a enzimas) es una técnica de inmuno-ensayo en la cual un antígeno inmovilizado se detecta mediante un anticuerpo enlazado a una enzima capaz de generar un producto detectable como cambio de color o algún otro tipo. La aparición de colorantes permite medir indirectamente mediante espectrofotometría el antígeno en la muestra. Es el examen utilizado para la detección del VIH en el ADN.
La Técnica de Sorthern se emplea para identificar secuencias específicas de DNA, mediante rompimiento con enzimas de restricción. Los fragmentos obtenidos se someten a electroforesis en un gel de agarosa, donde los fragmentos de mayor tamaño quedaran en la parte superior y las de menor tamaño en la parte inferior.
La técnica de Northen se emplea para identificar secuencias específicas de RNA. Consiste en aislarlo del citoplasma o del núcleo y se somete a electroforesis en gel de agarosa en condiciones desnaturalizantes para que los RNA se separen según su clase o peso molecular.
La electroforesis, mencionada en casi todas las técnicas anteriores, nos ayuda a observar los ácidos nucleicos y proteínas al final del procedimiento. En él las moléculas migran a través de un gel en el cual, por acción de un campo eléctrico, serán separadas de acuerdo a su peso molecular.
Jhomairy Miller 2012-1419
2013-0654
ResponderEliminar)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
2- Enzima de restricción (BRENDA)
Es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
Tema 1: REACCIÓN EN CADENA DE LA POLIMERASA (PCR)
ResponderEliminarEste método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde.
ETAPAS:
A) Desnaturalización: Se comienza calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrógeno y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos.
B)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.
Rocio Garcia .............2012-1235
ResponderEliminarReaccion de cadena polimerasa
La Reacción en Cadena de la Polimerasa (PCR) es una técnica "in vitro" que imita la habilidad natural de la célula de duplicar el ADN. Se trata de una técnica usada para crear un gran número de copias de un segmento de ADN, que utiliza ciclos de desnaturalización, apareamiento con cebadores y extensión por una ADN polimerasa termoresistente. El único método para obtener grandes cantidades de un fragmento de ADN era clonándolo en vectores adecuados e introduciéndolo y multiplicándolo en bacterias. En el año 1985, un investigador norteamericano, Kary Mullis (acreedor del Premio Nobel en Química 1993 por este aporte), desarrolló un método que permite, a partir de una muestra muy pequeña de ADN, obtener millones de copias de ADN in vitro, en unas pocas horas y sin necesidad de usar células vivas. Esta técnica, llamada reacción en cadena de la polimerasa (PCR), requiere conocer la secuencia de nucleótidos de los extremos del fragmento que se quiere amplificar. Estas secuencias se usan para diseñar dos oligonucleótidos sintéticos de ADN complementarios a una porción de cada una de las dos cadena de la doble hélice.
Tema 1: REACCIÓN EN CADENA DE LA POLIMERASA (PCR)
ResponderEliminarEste método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde. Al final del proceso se obtendrán 2 moléculas de ADN, cada una de ellas formada por una cadena original y una cadena complementaria que ha sido sintetizada. La enzima que realiza este proceso se denomina DNA polimerasa. Esta enzima añade los nucleótidos en el hidroxilo 3 terminal de la cadena de DNA paterna.
ETAPAS:
A) Desnaturalización: Se comienza calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrógeno y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación.
B)Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
Ray Cabrera 2013-0206
ResponderEliminartema 2- enzima de restricción (brenda)
es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de adn y cortar el adn en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
el mecanismo de corte de dna se realiza a través de la ruptura de dos enlaces fosfodiéster en la doble hebra, lo que da lugar a dos extremos de dna. éstos pueden ser romos (cuando los enlaces rotos coinciden) o cohesivos/escalonados. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
Ray Cabrera 2013-0206
ResponderEliminarTema 3 - ELISA…
El ensayo inmunoabsorbente ligado a enzimas (ELISA) utiliza como sus siglas lo indican una enzima como marcador para mediar la formación de complejos antígeno-anticuerpo.
Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario. Casi todas las pruebas ELISA son ensayos en fase sólida en los cuales se adsorbe un antígeno o un anticuerpo sobre un soporte sólido. Algunos de los protocolos se basan en reacciones de enlace competitivo y otras en reacciones de enlace no competitivo, pero en todas las pruebas ELISA se requiere de un paso de separación para eliminar el conjugado enzimático libre antes de proceder a determinar la cantidad de conjugado enzimático enlazado. Para lo cual se añade sustrato enzimático y se mide la reacción catalítica entre la enzima y el sustrato. Por sus características catalíticas las enzimas son marcadores muy sensibles y versátiles. Una sola proteína enzimática puede transformar en algunos minutos gran número de moléculas de sustrato en una cantidad igualmente abundante de producto final, produciendo un cambio de color amplificado y que se detecta con facilidad.En el método ELISA de inhibición el cambio de color se reduce, porque la actividad enzimática se inhibe cuando el anticuerpo se enlaza al conjugado enzimático.
Tema4 - Southern Blot para ADN…
Southernblot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda. Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern. El sector de ADN buscando puede ser un gen, o parte del mismo. Se puede utilizar para el diagnostico molecular de patologiasgeneticas. LA doble hebra de ADN se forma por complementariedad SONDA mustra (una de las hebras es la sonda en simple caden y la otra es la muestra en simple cadena). Los nucleotidos de la sonda esta macados P 32 (radioactivos); si al revelar hay marcador radioactivo presente, esta en la muestra analizada la secuencia complementaria a la sonda. Pasos: extraccion de ADN. Fragmentar el ADN en porciones mas pequeñas (enzimas de resctriccion). Separar los ragmentos realizando una corrida electroforetica usando como sustrato de migracion un gel. (separacion por tamaño). Se agrega la sonda sobre la membrana y se incuba. Se lava para quitar el maximo de hibridaciones inespecificas. Se revela por autoradiografia.
Ray Cabrera 2013-0206
ResponderEliminarHibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
C)Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
2- Enzima de restricción (BRENDA)
Es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
Ray Cabrera 2013-0206
ResponderEliminarTema 5-NORTHERNBLOT PARA RNA
Northernblot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente.
El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southernblot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northernblot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.
Ray Cabrera 2013-0206
ResponderEliminarTEMA 6-EASTERN BLOT
Se considera una extencion de la tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma.
pueden examinar la cantidad de proteínas en una muestra y comparar los niveles de presencia entre varios grupos. Otras técnicas ,también usando anticuerpos, permiten la detención de proteínas en tejidos (inmunohistoquímica) y células (inmunocitoquímica).
En el Northernblot, se utiliza para identificar las cadenas de ARN de secuencia semejante al ADN que se usa como sonda; a diferencia del tipo Southern que se utiliza para identificar ADN, el método northern sirve para identificar ARN.
Ray Cabrera 2013-0206
ResponderEliminarTEMA 7-WESTERN BLOT PARA PROTEINA
EL WESTERN BLOT ES UN MÉTODO EN BIOLOGÍA MOLECULAR/BIOQUÍMICA/INMUNOGENÉTICA PARA LA DETECCIÓN DE PROTEÍNAS EN UNA MUESTRA DE UN TEJIDO HOMOGENEIZADO O EXTRACTO. SE UTILIZA UNA ELECTROFORESIS PARA SEPARAR PROTEÍNAS DESNATURALIZADAS DE LA MASA. LAS PROTEÍNAS SON TRASFERIDAS DESDE EL GEL HACIA LA MEMBRANA (ORIGINALMENTE DE NITROCELULOSA), DONDE SON EXAMINADAS UTILIZANDO ANTICUERPOS ESPECÍFICOS PARA LA PROTEÍNA. COMO RESULTADO, SE PUEDEN EXAMINAR LA CANTIDAD DE PROTEÍNAS EN UNA MUESTRA Y COMPARAR LOS NIVELES DE PRESENCIA ENTRE VARIOS GRUPOS. OTRAS TÉCNICAS ,TAMBIÉN USANDO ANTICUERPOS, PERMITEN LA DETENCIÓN DE PROTEÍNAS EN TEJIDOS (INMUNOHISTOQUÍMICA) Y CÉLULAS (INMUNOCITOQUÍMICA).
EN EL NORTHERNBLOT, SE UTILIZA PARA IDENTIFICAR LAS CADENAS DE ARN DE SECUENCIA SEMEJANTE AL ADN QUE SE USA COMO SONDA; A DIFERENCIA DEL TIPO SOUTHERN QUE SE UTILIZA PARA IDENTIFICAR ADN, EL MÉTODO NORTHERN SIRVE PARA IDENTIFICAR ARN.
DE MANERA RESUMIDA, EL WESTERN SIRVE PARA IDENTIFICAR PROTEÍNAS Y EL NORTHERN PARA IDENTIFICAR SECUENCIAS DE RNA.
Ray Cabrera 2013-0206
ResponderEliminarTEMA8-SOUTHERN BLOT PARAADN Y PROTEINA
El SouthernBlot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizandosonicaciónó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAdsóRNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.
Ray Cabrera 2013-0206
ResponderEliminarTema 9- Extracción de adn mitocondrial
El ADN mitocondrial (mtDNA) es un material genético circular cerrado de doble cadena que se localiza en el interior de las mitocondrias celulares.El genoma mitocondrial consta de aproximadamente 16500 pares de bases (p.b.), y codifica para una pequeña fracción de las proteínas mitocondriales. El mtDNA contiene información de 38 genes: 2rRNA (12S y 16S), 22tRNA y 13 genes estructurales, los cuales codifican diferentes subunidades de los complejos enzimáticos del sistema de fosforilaciónoxidativa.La región mayor no codificante, conocida como región control o D-Loop, ocupa 1122 pares de bases.
Este comentario ha sido eliminado por el autor.
ResponderEliminarRay Cabrera 2013-0206
Eliminar8-Southern Blot para adn y proteina
El Southern Blot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizando sonicación ó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAds ó RNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.El traspaso de las muestras desde el gel al filtro es una etapa esencial, porque el reconocimiento debases complementarias entre sonda y secuencias de DNA en el gel es muy ineficiente.Cabe señalar que el nombre de la técnica Southern Blot deriva en parte del apellido Southern delinvestigador que la desarrolló y de la palabra en inglés Blot que significa traspasar
Ray Cabrera 2013-0206
ResponderEliminarTEMA 10- ELECTRORESIS DE PROTEINA
Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción
La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína.
La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL.
Este comentario ha sido eliminado por el autor.
ResponderEliminarclarianne NEvarez 2012-0740
ResponderEliminarMANEJOS DE MUESTRA Y REACTIVOS
La pureza de los reactivos es fundamental para la exactitud que se obtiene en cualquier análisis. En el laboratorio se dispone de distintos tipos de reactivos (sólidos, líquidos o disoluciones preparadas) tal y como se comercializan.
En general, las casas comerciales ofrecen un mismo producto con varias calidades. Es importante que cuando seleccionemos un reactivo su calidad esté en concordancia con el uso que se le va a dar.
2.1. Clasificación
En el laboratorio de análisis se utilizan reactivos de calidad analítica que se producen comercialmente con un alto grado de pureza. En las etiquetas de los frascos se relacionan los límites máximos de impurezas permitidas por las especificaciones para la calidad del reactivo o los resultados del análisis para las distintas impurezas. Dentro de los reactivos analíticos pueden distinguirse tres calidades distintas:
• Reactivos para análisis (PA): Son aquellos cuyo contenido en impurezas no rebasa el número mínimo de sustancias determinables por el método que se utilice.
• Reactivos purísimos: Son reactivos con un mayor grado de pureza que los reactivos “para análisis” .
• Reactivos especiales: Son reactivos con calidades específicas para algunas técnicas analíticas, como cromatografía líquida (HPLC), espectrofotometría (UV)…
Hay reactivos que tienen características y usos específicos como los reactivos calidad patrón primario, que se emplean en las técnicas volumétricas, o los patrones de referencia.
Clarianne Nevarez 2012-0740
ResponderEliminarREACCION EN CADENA DE POLIMERASA
La reacción en cadena de la polimerasa, conocida como PCR por sus siglas en inglés (polymerase chain reaction), es una técnica de biología molecular desarrollada en 1987 porKary Mullis,1 cuyo objetivo es obtener un gran número de copias de un fragmento de ADNparticular, partiendo de un mínimo; en teoría basta partir de una única copia de ese fragmento original, o molde.
Esta técnica sirve para amplificar un fragmento de ADN; su utilidad es que tras la amplificación resulta mucho más fácil identificar con una muy alta probabilidad, virus obacterias causantes de una enfermedad, identificar personas (cadáveres) o hacer investigación científica sobre el ADN amplificado. Estos usos derivados de la amplificación han hecho que se convierta en una técnica muy extendida, con el consiguiente abaratamiento del equipo necesario para llevarla a cabo.
Clarianne Nevarez 2012-0740
ResponderEliminarENZIMA DE RESTRICCIÓN
Un enzima de restricción (o endonucleasas de restricción) es aquella que puede reconocer una secuencia característica de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto, llamado sitio o diana de restricción, o en un sitio no muy lejano a este. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son reconocidos.
El mecanismo de corte de DNA se realiza a través de la ruptura de dos enlaces fosfodiéster en la doble hebra, lo que da lugar a dos extremos de DNA. Éstos pueden ser romos (cuando los enlaces rotos coinciden) o Cohesivos/escalonados. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
Los fragmentos de ADN obtenidos de este modo pueden unirse por otras enzimas llamadas ligasas.
Las enzimas de restricción que a pesar de ser distintas y provenir de distintas especies, tienen la misma secuencia de reconocimiento y dejan el mismo extremo cohesivo, pero no cortan en el mismo sitio, son llamadas isoesquizómeros. Por ejemplo, están los isoesquizómeros Asp718 y KpnI.
El Premio Nobel de Medicina de 1978 fue concedido a los microbiólogos Werner Arber, Daniel Nathans y Hamilton Smith por el descubrimiento de las endonucleasas de restricción lo que condujo al desarrollo de la tecnología de ADN recombinante. El primer uso práctico de su trabajo fue la manipulación de la bacteria E. coli para producir insulina humana para los diabéticos.
Uno de los campos en los que las enzimas de restricción han tenido mayor implicación ha sido el diagnóstico de enfermedades genéticas relacionadas con cambios en la secuencia del ADN, ya sean mutaciones puntuales, inserciones o deleciones de fragmentos. Si éstas se producen en un sitio de reconocimiento de la enzima de restricción, al producirse eliminarán o agregarán nuevos sitios de corte. Al aplicar esta enzima al gen de una persona sana y una enferma se deberían observar distintas cantidades de fragmentos para cada caso en una electroforesis.
Clarianne Nevarez 2012-0740
ResponderEliminarElisa Directo Samdwis
ELISA (acrónimo del inglés Enzyme-Linked ImmunoSorbent Assay, Ensayo por inmunoabsorción ligado a enzimas) es una técnica de inmunoensayo en la cual un antígeno inmovilizado se detecta mediante un anticuerpo enlazado a una enzima capaz de generar un producto detectable como cambio de color o algún otro tipo; en ocasiones, con el fin de reducir los costos del ensayo, nos encontramos con que existe un anticuerpo primario que reconoce al antígeno y que a su vez es reconocido por un anticuerpo secundario que lleva enlazado la enzima anteriormente mencionada. La aparición de colorantes permite medir indirectamente mediante espectrofotometría el antígeno en la muestra.
Se usa en muchos laboratorios para determinar si un anticuerpo particular está presente en la muestra de sangre de un paciente. Aunque el procedimiento es rutinario y sencillo, involucra a un gran número de variables, tales como seleción de reactivo, temperatura, medición de volumen y tiempo, que si no se ajustan correctamente puede afectar a los pasos sucesivos y al resultado de la prueba.
La interacción antígeno-anticuerpo en el laboratorio puede ser utilizada para determinar si un paciente tiene una infección o una enfermedad autoinmune. Pero como prueba diagnóstica, posee diversas limitaciones que conviene conocer:
-En primer lugar, un resultado positivo que confirma la presencia de anticuerpos no significa necesariamente que el paciente esté enfermo. El cuerpo de una persona que ha estado enferma y que ya se ha recuperado, puede seguir produciendo anticuerpos. Esto originaría un falso positivo.
-En segundo lugar, hay personas que producen una baja cantidad de anticuerpos, por lo que éstos pueden pasar desapercibidos y no ser medidos, dando lugar a un falso negativo. Sería el caso, por ejemplo, de personas que padezcan una inmunodeficiencia, o que se encuentren en el periodo ventana de la infección en el momento de realizar la prueba, o que estén infectadas por una cepa extraña.
-En tercer lugar, pueden aparecer falsos positivos cuando se da inespecificidad entre antígeno-anticuerpo.
En estos casos de diagnóstico de enfermedad es recomendable la eliminación (mediante centrifugación) de células de la sangre que puedan interferir con el ensayo y pueda ocasionar un resultado falso positivo, careciendo aquél de especificidad. También, debemos tener en cuenta que cuando se trata de diagnosticar una enfermedad que posee un valor predictivo positivo bajo en una determinada población, es decir, que esa enfermedad tiene muy baja incidencia en dicha población, es necesario volver a confirmar el resultado positivo mediante otro método de diagnóstico independiente. Normalmente, se lleva a cabo un western blot donde se detecta la presencia de varios anticuerpos, simultáneamente, frente a la misma infección en una muestra. El resultado del western se considera positivo cuando aparecen al menos 5 bandas, que indica que 5 anticuerpos diferentes están presentes en el sujeto frente a esa infección. Entonces es cuando se diagnostica como positivo a dicho paciente.
Clarianne Nevarez 2012-0740
ResponderEliminarTEMA :Elisa Indirecto
La técnica de ELISA es un ensayo inmunoenzimático ampliamente empleado en el área médica para la cuantificación de moléculas especialmente de aquellas que experimentan cambios en diferentes estados como pueden ser infecciones por bacterias, virus, hongos o parásitos o fases activas de enfermedades autoinmunes. Como ejemplo se pueden medir por ELISA hormonas, autoanticuerpos, inmunoglobulinas contra antígenos de patógenos, toxinas, antígenos. Las técnicas de ELISA son también muy usadas en los ensayos de nuevos medicamentos para comprobar sus efectos cuantificando las moléculas de interés en cada caso. Existen numerosas variantes de este tipo de ensayo. Entre ellos se encuentran los métodos directo e indirecto. El método directo permite la detección del antígeno con un anticuerpo específico conjugado con una enzima como sistema de marcaje. En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA.
Clarianne Nevarez 2012-0740
ResponderEliminarTEMA 4 :Southern blot,
Southern blot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda.1 Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern.
1. Extracción del ADN
Se puede extraer ADN de casi cualquier tejido humano. Las posibles fuentes de ADN en la escena de un delito incluyen sangre, semen, tejido de una víctima muerta, células del folículo capilar y saliva. El ADN extraído de las pruebas del delito ("indicios" o "vestigios" biológicos) se compara con el extraído de muestras de referencia, obtenidas de personas conocidas,habitualmente de la sangre.
2. Digestión del ADN con una endonucleasa de restricción
El ADN extraído de la muestra se trata con una endonucleasa de restricción,que es una enzima que corta el ADN bicatenario en donde tenga una secuencia característica. La enzima que se usa más frecuentemente para el análisis legal es HaeIII, que corta el ADN en la secuencia 5'-GGCC-3'.
3. Electroforesis en gel de agarosa
Tras la digestión del ADN, los fragmentos de ADN resultantes se separan según su tamaño mediante electroforesis en geles de agarosa. Durante la electroforesis, las moléculas de ADN, que poseen carga negativa, migran hacia el electrodo positivo. Al avanzar las moléculas de ADN, su velocidad de migración se ve reducida por la matriz del gel de agarosa. Las moléculas menores se mueven más deprisa a través de los poros del gel que las de mayor tamaño. Como resultado, se produce una separación continua de los fragmentos de ADN de acuerdo con su tamaño, de modo que los fragmentos más pequeños avanzan la mayor distancia con referencia al origen o punto de aplicación de la muestra.
4. Preparación de un ensayo de Southern ("Southern blot")
Tras la electroforesis, las moléculas de ADN separadas se desnaturalizan mientras permanecen en el gel de agarosa, impregnando éste con una disolución alcalina. Tras la neutralización de ésta, el DNA monocatenario resultante se transfiere a la superficie de una membrana de nailon,realizando así una copia o "calco" (la traducción más literal del inglés blot,conservando este sentido, es "secante"). Este proceso de desnaturalización y transferencia se conoce como método de Southern en recuerdo de quien lo inventó, Edward Southern. Al igual que la aplicación de un secante a un papel con la tinta húmeda transfiere una réplica de la imagen del papel al secante,el "calco" del ADN en el gel a la membrana de nailon conserva la distribución espacial de los fragmentos de ADN conseguida en el gel como resultado de la electroforesis.
Clarianne Nevarez 2012-0740
ResponderEliminarTEMA 5 :NORTHERN BLOT
Northern blot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente.1
El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southern blot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northern blot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.
PROCEDIMIENTO
El procedimiento general comienza con la extracción del RNA total de una muestra de tejido homogenizado. El mRNA se puede aislar a través del uso de cromatografía para mantener solamente los ARN con colas de poli-A. Las muestras de ARN son entonces separadas por electroforesis en gel. Dada la fragilidad de los geles y la incapacidad de las sondas para penetrar en la matriz, las muestras de RNA, separadas por tamaño tras la electroforesis, serán tranferidas a una membrana de Nailon por medio de capilaridad o un sistema de transferencia al vacío.
Los sistemas de capilaridad iónica se utilizan pra transferir el ARN desde un gel de electroforesis a una membrana de Northern blot.
Una membrana de Nylon cargada positivamente es el soporte mas efectivo para usar en el northern blot ya que los ácidos nucleicos, que están cargados negativamente, tienen una alta afinidad por estas membranas. El buffer de transferencia usado para el ensayo suele contener formamida, dado que ésta consigue disminuir la temperatura de hibridación de la sonda, lo cual previene la degradación del ARN por las altas temperaturas. Una vez que el ARN ha sido transferido a la membrana, este es inmovilizado a través de enlaces covalentes formados con la membrana, lo cual se consigue por medio de luz ultravioleta o calor. Después del marcaje de la sonda, ésta se hibrida con el RNA en la membrana. Las condiciones experimentales que pueden afectar la eficiencia y especificidad de la hibridación están determinadas por las condiciones iónicas, de viscosidad, la presencia de dobles hebras de RNA, bases desemparejadas y composición de las mismas. Finalmente, la membrana debe de lavarse para asegurar que la sonda se ha unido de forma específica y para evitar ruido de fondo en las señales emitidas por la misma. Estas señales serán detectadas por Rayos X y pueden ser cuantificadas mediante técnicas de densitometría. Para crear controles y poder asegurarnos de que no se están mostrando genes que no nos interesen se puede realizar posteriormente la determinación por microarrays o RT-PCR.
Clarianne Nevarez 2012-0740
ResponderEliminarTEMA 6-EASTERN BLOT
Se considera una extencion de la tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma.
pueden examinar la cantidad de proteínas en una muestra y comparar los niveles de presencia entre varios grupos. Otras técnicas ,también usando anticuerpos, permiten la detención de proteínas en tejidos (inmunohistoquímica) y células (inmunocitoquímica).
En el Northernblot, se utiliza para identificar las cadenas de ARN de secuencia semejante al ADN que se usa como sonda; a diferencia del tipo Southern que se utiliza para identificar ADN, el método northern sirve para identificar ARN.
Clarianne Nevarez 2012-0740
ResponderEliminarTEMA 7 :El Western blot, o inmunoblot, es una técnica analítica usada para detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis en gel se separan las proteínas atendiendo al criterio que se desee: peso molecular, estructura, hidrofobicidad, etc. Hay casi tantas posibilidades como tipos de electroforesis existen. Luego son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella.1 2 Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas.
Esta técnica es hoy en día imprescindible en varios campos de la biología, como la biología molecular, la bioquímica, la biotecnología o la inmunología. Existe toda una industria especializada en la venta de anticuerpos (monoclonales y policlonales) contra decenas de miles de proteínas diferentes.3 4
El Western blot fue desarrollado en el laboratorio de George Stark, en la Universidad de Stanford.2 El nombre (Western, occidental en inglés) le fue dado por W. Neal Burnette,5 y consiste en un juego de palabras con una técnica análoga pero que usa DNA, el Southern (sureño) blot, que en este caso debe su nombre a su descubridor, Edwin Southern. Otras técnicas que fueron nombradas siguiendo este criterio son el Northern blot (en el que se separa e identifica RNA), el Eastern blot, el Southwestern blot.
Preparación de la muestra
Las muestras pueden ser tomadas de un tejido entero o de un cultivo celular:
Una pequeña cantidad del tejido se introduce en un buffer de extracción. Después se procede a homogeneizarlos, sea con una licuadora (para grandes volúmenes de muestra), con un homogenizador (para muestras pequeñas) o por sonicación. Por último se centrifuga para obtener las proteínas en el sobrenadante, la fracción no precipitada.6
Las células pueden lisarse por uno de esos métodos mecánicos. También pueden usarse detergentes, sales o tampones que favorezcan la lisis y solubilicen las proteínas. A veces se añaden inhibidores de proteasas y fosfatasas que evitan la digestión por las enzimas celulares de las proteínas y de las fosfoproteínas, respectivamente.7
Los materiales deben estar a bajas temperaturas (~4 °C) para evitar la desnaturalización proteica. Para separar proteínas de compartimentos y orgánulos celulares es preciso combinar técnicas mecánicas - como filtraciones y centrifugaciones - y bioquímicas.
Clarianne Nevarez 2012-0740
ResponderEliminarTEMA 9:LA TÉCNICA DE TRANSFERENCIA BLOTTING
La técnica de transferencia (blotting) se basa en el mismo fundamento que la autorradiografía, y permite identificar la presencia de una banda concreta en un gel de electroforesis, bien sea de proteínas o de ácidos nucleicos:
Si se trata de proteínas, la técnica se llama Western blot. Se utilizan anticuerpos marcados con radioactividad para identificar cuál es la banda que presenta la proteína que me interesa localizar
Si se trata de DNA, la técnica se llama Southern blot. Se utilizan sondas de DNA (moléculas de DNA marcadas con radioactividad y con una secuencia conocida) para identificar qué bandas del gel contienen secuencias complementarias a la de la sonda
Si se trata de RNA, la técnica se llama Northern blot. Se utilizan sondas de RNA (moléculas de RNA marcadas con radioactividad y con una secuencia conocida) para identificar qué bandas del gel contienen secuencias complementarias a la de la sonda
Clarianne Nevarez 20120740
ResponderEliminarTEMA10: ELECTROFORESIS DE PROTEÍNAS EN SUERO
Este examen mide los tipos de proteína en la parte líquida (suero) de una muestra de sangre.
Ver también:
Forma en que se realiza el examen
Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción
La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína.
La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL).
PREPARACIÓN PARA EL EXAMEN
A usted le pueden solicitar no comer ni beber nada durante 12 horas antes de un examen de electroforesis de lipoproteínas.
El médico le puede solicitar que deje de tomar medicamentos que pudieran afectar el examen. NO suspenda ningún medicamento sin consultarlo previamente con éste.
Los medicamentos que pueden afectar la medición de las proteínas totales incluyen clorpromazina, corticoesteroides, isoniazida, neomicina, fenacemida, salicilatos, sulfamidas y tolbutamida.
LO QUE SE SIENTE DURANTE EL EXAMEN
Cuando se inserta la aguja para extraer la sangre, algunas personas sienten un dolor moderado, mientras que otras sólo sienten un pinchazo o sensación de picadura. Posteriormente, puede haber algo de sensación pulsátil.
RAZONES POR LAS QUE SE REALIZA EL EXAMEN
Las proteínas están compuestas de aminoácidos y son componentes importantes de todas las células y los tejidos. Existen muchas clases de proteínas en el cuerpo, con muchas funciones diferentes. Los ejemplos de proteínas son: las enzimas, algunas hormonas, la hemoglobina, la lipoproteína de baja densidad (colesterol "malo") y otras.
Las proteínas séricas se clasifican como albúmina y globulinas. La albúmina es la proteína de mayor concentración en el suero. Trasporta muchas moléculas pequeñas, pero también es importante para impedir que el líquido se escape de los vasos sanguíneos hacia los tejidos.
Las globulinas se dividen en alfa-1, alfa-2, beta y gammaglobulinas. En general, los niveles de las proteínas alfa y gammaglobulinas aumentan cuando hay inflamación en el cuerpo.
2012-1330
ResponderEliminarTema 2 :Reacción de cadena polimerasa (PCR)
La reacción en cadena de la polimerasa, ya más conocida como PCR, es una técnica que permite replicar entre cientos de miles y millones de veces, en el transcurrir de pocas horas e in vitro, pequeñas cantidades de ADN. Uno de los aportes fundamentales de esta metodología a la investigación básica en biología molecular consiste, precisamente, en la rapidez y eficiencia mediante las cuales se realiza una tarea que antes requería largas y tediosas jornadas. El producto quese obtiene al finalizar la reacción es una gran cantidad de un fragmento génico con alto grado de pureza- favorece la tarea de los investigadores empeñados en ampliar nuestros conocimientos sobre la estructura y función de los genes. El método se basa, en su forma más simple en la realización de tres reacciones sucesivas llevadas a cabo a distintas temperaturas. Estas reacciones se repiten cíclicamente entre veinte y cuarenta veces (véase la figura 1). La muestra se calienta, en el primer paso, hasta lograr la separación de las dos cadenas que constituyen el ADN, hecho que se conoce como "desnaturalización". En el segundo paso, la temperatura se reduce para permitir el "apareamiento" de cada una de dos cadenas cortas de nucleótidos (oligonucleóridos) con cada una de las hebras separadas del ADN molde. Se trata de segmentos de ADN de cadena simple, sintetizados en el laboratorio y diseñados de manera tal que permiten definir los límites del tramo de ADN que se desea replicar. Obviamente, para que se pueda producir el apareamiento, cada uno de estos oligonucleótidos, a los que se denomina "iniciadores" o primers, debe ser complementario del tramo al que tienen que unirse en las cadenas separadas del ADN molde. En tercer lugar, una enzima ADN polimerasa extiende los primers, en el espacio comprendido entre ambos, sintetizando las secuencias complementarias de las hebras del ADN molde. Para ello, la ADN polimerasa usa desoxidonucleósidos trifosfato agregados a la mezcla de reacción. La temperatura a la que se realiza el tercer paso está condicionada por aquélla a la cual "trabaja" la enzima ADN polimerasa. Al cabo del primer ciclo de tres reacciones (desnaturalización, apareamiento, extensión) el tramo de ADN elegido se ha duplicado y el doble de su cantidad original se encuentra disponible para ser nuevamente replicado en un segundo ciclo. El resultado de la aplicación de numerosos ciclos "en cadena" da lugar a la amplificación geométrica del segmento de ADN delimitado por los primers.Para replicar el ADN, la técnica PCR hizo uso, en un principio, de la ADN polimerasa de la bacteria Escherichia coli. Pero esta enzima resulta desactivada debido a la alta temperatura requerida para desnaturalizarla doble cadena del ADN, por lo cual debía agregarse enzima fresca al comenzar el tercer paso de cada ciclo. Este inconveniente fue solucionado de manera ingeniosa cuando se la reemplazó por su equivalente de la bacteria "termófila" Thermus aquaticus. En efecto, la ADN polimerasa de este microrganismo, denominada Taq polimerasa, actúa eficientemente entre los 75° C y los 80° C y resiste más de dos horas a 93° C. De esta manera es posible mezclar todos los componentes de la PCR al comenzar el primer ciclo y la reacción en cadena puede llevarse a cabo mediante equipos automatizados que realizarán los ciclos térmicos programados.
2012-1330
ResponderEliminartema 3: Enzimas de restrincion (BRENDA)
Las enzimas de restricción son proteínas cuya función es cortar las hebras de ADN. Se podría decir que son “tijeras moleculares” que cortan ADN. Lo hacen en forma específica. Esto significa que cada enzima reconoce un sitio particular del ADN, es decir que reconoce una secuencia particular de nucleótidos. Esa secuencia específica para cada enzima se denomina “sitio de restricción”. Una vez que la enzima reconoce estos sitios, se posiciona sobre la molécula de ADN y corta dentro o en torno de esa secuencia. Las enzimas de restricción, conocidas también como endonucleasas, sólo cortan el ADN si reconocen en su interior una secuencia específica de nucleótidos. Estas enzimas fueron descubiertas en microorganismos. De hecho, se encuentran sólo en organismos procariotas (bacterias). Por esto, se les dio una nomenclatura asociada al organismo de donde provienen. Por ejemplo, las enzimas de restricción que se descubrieron en la bacteria Escherichia coli se denominan Eco. Existen diferentes tipos de enzimas Eco que se diferencian en la secuencia que reconocen y cortan. Para diferenciarlas se les agregan letras y números romanos, por ejemplo: Eco RI (Eco “erre” “uno”). Así, el sitio de restricción para EcoRI es la secuencia GAATTC, como muestra la ilustración anterior. Una vez que la enzima encontró ese sitio en el ADN, se acerca a la hebra de ADN y realiza el corte entre la G y la A. Al investigar otras especies de bacterias se descubrieron cientos de enzimas de restricción distintas y cada una reconoce una región específica. Estas enzimas fueron descubiertas en la década del ’70 y hasta la fecha existen más de 250 enzimas de restricción. Se cree que la función natural de estas enzimas en las bacterias es protegerlas contra ADN de virus que podrían ingresar en sus células. De esta manera, la bacteria utiliza estas “tijeras moleculares” para cortar en pedacitos el ADN viral que la infecta. El ADN propio de la bacteria no se corta, pues lo tiene “protegido” contra sus propias enzimas de restricción. Usos de las enzimas de restricción
Las enzimas de restricción tienen diferentes aplicaciones que son de gran importancia en investigaciones en biología molecular y en las técnicas que emplea la biotecnología moderna:
1. Hacer mapa de restricción de un plásmido o bacteriófago. El ADN se corta con varias enzimas de restricción, solas y en parejas, para determinar el número de sitios de corte y sus posiciones relativas en la molécula, el orden y la distancia entre ellos.
2. Fragmentar ADN para separación por electroforesis. Los fragmentos obtenidos después de la actuación de las distintas enzimas de restricción, se pueden separar por tamaños mediante la técnica de electroforesis y así estudiar los distintos fragmentos. Por ejemplo: para la técnica de Southern blotting o en las usadas para identificar polimorfismos de ADN en distintos individuos .
3. Generación de fragmentos para ser clonados en los vectores apropiados, y crear ADN recombinante. Se puede cortar una molécula de ADN con una enzima y, con el mismo tipo de enzima, cortar el fragmento de ADN de interés para clonar. Se unen con ligasas estas dos moléculas de ADN, generando así una molécula de ADN recombinante. Este vector recombinante puede usarse para transformar células que expresen el gen de interés clonado (si se usó un vector de expresión con el promotor adecuado) o puede usarse simplemente para tener clonado (“guardado”) ese fragmento de ADN de interés. Por ejemplo: para los proyectos de secuenciación de genomas
2012-1330
ResponderEliminartema 4: ELISA directo (Sandwish)
La técnica de ELISA (Enzyme Linked InmunoSorbent Assay) es un procedimiento de ensayo inmuno enzimático. Se basa en la detección antígeno(Ag) o anticuerpo (Ac), La técnica de ELISA es un ensayo inmunoenzimático ampliamente empleado en el área médica para la cuantificación de moléculas especialmente de aquellas que experimentan cambios en diferentes estados como pueden ser infecciones por bacterias, virus, hongos o parásitos o fases activas de enfermedades autoinmunes. Como ejemplo se pueden medir por ELISA hormonas, autoanticuerpos, inmunoglobulinas contra antígenos de patógenos, toxinas, antígenos. Las técnicas de ELISA son también muy usadas en los ensayos de nuevos medicamentos para comprobar sus efectos cuantificando las moléculas de interés en cada caso. Existen numerosas variantes de este tipo de ensayo. Entre ellos se encuentran los métodos directo e indirecto. El método directo permite la detección del antígeno con un anticuerpo específico conjugado con una enzima como sistema de marcaje. Técnica: Las placas ELISA se preparan recubriendo los pocillos con las soluciones en las que se sospecha se encuentra el antígeno. Se incuban con anticuerpos marcados. Indican la presencia de antígeno en la solución analizada. Es necesario incluir controles negativos que serán muestras del mismo tipo de las analizadas (sangre, orina) pero en las que se tenga la certeza de la ausencia del antígeno buscado. Asimismo se incluyen controles positivos (soluciones donde se encuentra el antígeno buscado).
Elisa sandwish Se trata de un ensayo muy empleado en el que se recubre el pocillo con un primer anticuerpo anti-antígeno. Después de lavar el exceso de anticuerpo se aplica la muestra problema en la que se encuentra el antígeno, que será retenido en el pocillo al ser reconocido por el primer anticuerpo. Después de un segundo lavado que elimina el material no retenido se aplica una solución con un segundo anticuerpo anti-antígeno marcado. Así pues cada molécula de antígeno estará unida a un anticuerpo en la base que lo retiene y un segundo anticuerpo, al menos, que lo marca. Este ensayo tiene una gran especificidad y sensibilidad debido a la amplificación de señal que permite el segundo anticuerpo.
2012-1330
ResponderEliminartema 5:ELISA Indirecto
En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA.
Es el ensayo más popular, como lo es la inmunofluorescencia indirecta, pues un mismo secundario marcado y un mismo sistema enzimático permite cuantificar una gran variedad de antígenos, por eso es un método más polivalente y barato, aunque se pierda algo de precisión por tener un eslabón más con respecto al método directo. La dilución de la solución que contiene el anticuerpo primario (por ejemplo: suero sanguíneo) es un factor muy importante a tener en cuenta para evitar la aparición de falsos negativos, ya que si la muestra está muy diluida nosaldrá positiva si la titulación de anticuerpos es muy baja. Es decir, aunque los anticuerpos están presentes, la prueba no da positivo porque la concentración de anticuerpos específicos contra el antígeno que está pegado en el fondo del pocillo no es suficiente como para dar una señal detectable.El ELISA indirecto es el método de elección para detectar la presencia de anticuerpos séricos contra el virus de la inmunodeficiencia humana (VIH), agente causante del síndrome de inmunodeficiencia adquirida (SIDA). Según esta técnica, proteínas recombinantes de la envoltura y el núcleo del VIH se absorben como antígenos en fase sólida a los pocillos. Las personas afectadas de VIH producen anticuerpos séricos contra epítopos en estas proteínas víricas. En general, el ELISA indirecto permite detectar anticuepos séricos contra VIH desde las primeras seis semanas de una infección.
2012-1330
ResponderEliminartema 6:Tecnicas Blotting
El término “blotting” hace referencia a la transferencia de macromoléculas biológicas desde un gel hasta una membrana y su posterior detección en la superficie de la misma. Este proceso es necesario ya que todas estas macromoléculas están embebidas dentro de la matriz que forma el gel, lo que imposibilita realizar su detección, mientras que en la membrana, se encuentran accesibles al estar adheridas sobre la superficie de la misma. La técnica del Western Blot (también llamado “inmunoblotting” debido a que se utilizan anticuerpos para detectar el antígeno o los antígenos específicos de interés) fue descrita por primera vez por Towbin et al. en 1979 y en la actualidad es una técnica de rutina en todos los laboratorios que realizan análisis de proteínas. La especificidad de la unión antígeno – anticuerpo permite la detección de una única proteína dentro de una mezcla compleja de otras proteínas. En la actualidad se utiliza como criterio de identificación positivo de una proteína especifica en una mezcla compleja y para obtener datos cualitativos y semicuantitativos sobre la misma.
El primer paso de todo Western Blot es la separación de las macromoléculas mediante geles de electroforesis; después de la misma, las macromoléculas ya separadas en función de su diferente peso molecular se transfieren a una segunda matriz, generalmente una membrana de nitrocelulosa o PVDF. Posteriormente, se bloquea la membrana para evitar la unión inespecífica a su superficie de los anticuerpos que se van a utilizar para la detección de la proteína de interés. En el siguiente paso, se une a dicha proteína transferida un anticuerpo especifico marcado con una enzima y, finalmente, se añade un sustrato apropiado para dicha enzima con lo que se produce un producto detectable como, por ejemplo, un precipitado cromogénico o fluorogénico en la membrana. En la actualidad, los métodos más sensibles emplean sustratos quimioluminiscentes que cuando se combinan con la enzima correspondiente, producen luz como producto final, que puede detectarse mediante una película o una cámara CCD. En todos los casos y sea cual sea el sustrato que se utilice, la intensidad de la señal se correlaciona con la cantidad del antígeno en la superficie de la membrana.
Este comentario ha sido eliminado por el autor.
ResponderEliminar2012-1330
ResponderEliminartema 7:Electroforesis de proteínas
Es un método de separación de proteínas mediante la aplicación de un campo eléctrico. Existen diferentes tipos en función del tipo de separación empleado: electroforesis de zona (separación en función de la carga), isoeléctrico enfoque y separación por tamaño en tamiz molecular (también aplicable a ácidos nucleicos). Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina).Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (-) o cátodo (+) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración.
Isoelectroenfoque: en lugar de separar las proteínas en función de su carga a un pH dado, se separan en función de su punto isoeléctrico (pI): el pI es el pH en el que la carga neta de la proteína es nula, y depende de la composición aminoacídica de la proteína. Se crea un gradiente de pH mediante anfolitos (estabilizan el pH a lo largo del gel). Cada proteína migrará hasta alcanzar su pI, punto en el cual precipitará al acumularse (de ahí el nombre, isoelectroenfoque).También se suelen utilizar acoplados a zimogramas si deseamos determinar el pI de una enzima o en electroforesis bidimensional como primera dimension.
Separación por tamaño: permite separar proteínas y ácidos nucleicos. En el caso de las proteínas, deben ser tratadas con SDS (sodio dodecil sulfato) para que su carga sea negativa y todas migren hacia el ánodo (no es necesario hacer eso con los ácidos nucleicos, ya que tienen carga negativa); la separación se hace en medios de soporte en el que se ha creado un tamiz molecular (matriz), que hace que las proteínas más pequeñas corran más que las más grandes.Una vez separadas las proteínas, deben fijarse y teñirse para poder ser visualizadas. Hay diferentes protocolos en función de lo que se quiere estudiar: fijación por calor o química y tinción en caso de estudios no específicos (proteinograma y electroforesis Hb, por ejemplo) o fijación mediante anticuerpos previa a la tinción en caso de estudiar proteínas específicas (inmunofijación).Por lo general para la tinción de geles de poliacrilamida se utiliza tinción con plata, tinción de Coomassie o tinción fluorescente. Dependiendo del fin de nuestro análisis. La tinción con plata no es utilizada si se desean hacer análisis por espectrometría de masas (MS) pero es más sensible que la tinción con Azul de Coomassie, por otro lado, la tinción fluorescente es muy sensible y compatible con MS pero los costos de tinción son elevados.Una tinción sensible y barata es la denominada "Blue Silver" o Coomassie G250.1 Además, esta tinción es compatible con MS. Está compuesta por azul de coomassie diluido en una solución coloidal y se utiliza agua destilada para destiñir el gel de poliacrilamida.
2012-1330
ResponderEliminartema 8:El ADN mitocondrial
Es un fragmento de ácido nucleico encontrado dentro de las mitocondrias. Las mitocondrias son pequeños organelos o estructuras celulares que se encuentran dentro de las células eucarióticas (generalmente las células animales). Estos organelos se usan básicamente para la producción y generación de energía para que la célula lleve a cabo sus funciones. Las plantas tienen cloroplastos. Las células animales tienen mitocondrias. Y cada una de estas mitocondrias tiene su propio genoma. Su propia molécula circular de ADN. Y eso es el ADN mitocondrial, porque en realidad es diferente del ADN que se encuentra en el núcleo de las células. No se encuentra dentro del núcleo. El número de genes en el ADN mitocondrial es de 37, frente a los 20.000 - 25.000 genes del ADN cromosómico nuclear humanos.Codifica dos ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa. El ADN mitocondrial está en replicación constante, independientemente del ciclo y del tipo celular. Se piensa que tiene lugar de forma asíncrona, es decir, que tiene lugar en las dos cadenas en tiempos diferentes y con dos orígenes distintos hacia direcciones contrarias. El comienzo tendría lugar en el origen de la cadena pesada, situado en el bucle D, y replicaría ésta tomando como molde la cadena ligera. Cuando se alcanza el segundo origen, situado a dos tercios de distancia del primero, comienza la segunda ronda de replicación en sentido opuesto. Se ha propuesto un nuevo sistema de replicación que coexistiría con el primero. Sería bidireccional y comportaría una coordinación entre hebras directas y retrasadas.
Tradicionalmente se ha considerado que el ADN mitocondrial humano se hereda sólo por vía materna. Según esta concepción, cuando un espermatozoide fecunda un óvulopenetra el núcleo y su cola junto con sus mitocondrias son destruidos en el óvulo materno. Por lo tanto, en el desarrollo del cigoto sólo intervendrían las mitocondrias contenidas en el óvulo. Sin embargo, se ha demostrado que las mitocondrias del espermatozoide pueden ingresar al óvulo. Según algunos autores el ADN mitocondrial del padre puede perdurar en algunos tejidos, como los músculos.Según otros, no llega a heredarse al ser marcado por ubiquitinación y degradado. El ADN mitocondrial puede ser usado para identificar individuos junto con otra evidencia. También es usado por laboratorios forenses para caracterizar viejas muestras de esqueleto humano. Distinto que el ADN nuclear, el ADN mitocondrial no sirve para identificar individuos sin ambigüedad, pero si para detectar parentescos entre grupos de individuos; es usado entonces para comparaciones entre personas desaparecidas y restos no identificados y sus familiares. El ADN mitocondrial humano tiene características únicas que lo hacen muy apropiado para estudios microevolutivos: la herencia del genoma mitocondrial se realiza exclusivamente por la vía materna, sin recombinarse; hay un fragmento en este genoma de 400pb (pares de bases) altamente polimórfico, y posee una alta frecuencia de mutaciones (5 a 10 veces mayor que el ADN nuclear)10 Este ADN se puede extraer de muestras de cualquier tejido, incluso de la sangre y del tejido óseo. Gracias a su presencia en el hueso se puede obtener el genoma de individuos ya muertos desde hace muchos años. El análisis de la secuencia genómica se usa para estudiar las relaciones filogenéticas, no sólo en humanos sino, también en muchos otros organismos. Por este motivo se utiliza para determinar variabilidad en poblaciones naturales (para ver si hay o no endogamia), información útil para la conservación de especies en peligro de extinción.
2012-1330
ResponderEliminartema 9;Anticuerpos monoclonales
Son anticuerpos homogéneos producido por una célula híbrida producto de la fusión de un clon de linfocitos B descendiente de una sola y únicacélula madre y una célula plasmática tumoral. Los anticuerpos monoclonales mAB, de la frase en inglés con idéntico significado que en español: monoclonal antibody, son anticuerpos idénticos porque son producidos por un solo tipo de célula del sistema inmune, es decir, todos los clones proceden de una sola célula madre. Es posible producir anticuerpos monoclonales que se unan específicamente con cualquier molécula con carácter antigénico. Este fenómeno es de gran utilidad en bioquímica, biología molecular y medicina. Si una sustancia extraña (antígeno) se inyecta en el cuerpo de un ratón o un humano, alguna de las células B de su sistema inmune se transformarán en células plasmáticasy empezarán a producir anticuerpos que se unirán a ese antígeno. Cada célula B produce un solo tipo de anticuerpo, pero diferentes linfocitos B producirán anticuerpos estructuralmente diferentes que se unen a distintas partes del antígeno. Esta mezcla fisiológica natural de anticuerpos es conocida como Antisuero policlonal.Para producir anticuerpos monoclonales, primero se extraen células B del bazo de un animal que ha sido expuesto al antígeno. Estas células B son fusionadas en presencia de PEG (polietilenglicol) con células tumorales de mieloma múltiple (un tipo de cáncer) que pueden crecer indefinidamente en cultivo celular. Esta fusión hace a las membranas celulares más permeables. Estas células fusionadas híbridas, llamadas hibridomas pueden multiplicarse rápida e indefinidamente, puesto que son células tumorales después de todo y pueden producir gran cantidad de anticuerpos. Los hibridomas son suficientemente diluidos y cultivados para obtener un número diferente de determinadas colonias, las cuales producen sólo un tipo de anticuerpo. Los anticuerpos de diferentes colonias son analizados para conocer su capacidad de unirse a un antígeno determinado, por ejemplo con un tipo de test llamado ELISA, y para seleccionarse y aislarse de la manera más efectiva.
Nuestro sistema inmune reconoce y ataca elementos ajenos al organismo, como bacterias causantes de enfermedades, virus y otros agentes infecciosos. Este sistema de defensa está basado en células especializadas y proteínas que protegen el organismo. Estas proteínas especializadas incluyen anticuerpos y nuestro organismo produce una enorme variedad de anticuerpos para ser capaz de interaccionar con prácticamente todo posible patógeno. Los anticuerpos tienen dos características muy útiles. En primer lugar, son extremadamente específicos, es decir, cada anticuerpo se une y ataca un único antígeno. En segundo lugar, algunos anticuerpos, una vez activados por la presencia de la enfermedad, continúan confiriendo resistencia contra esa enfermedad; ejemplos clásicos son los anticuerpos de las enfermedades de la infancia.
Kery Joussett Sanchez Alvarez 2012-0919
ResponderEliminarPodriamos decir que la La transcripción del ADN es el primer proceso de la expresión génica, mediante el cual se transfiere la información contenida en la secuencia del ADN hacia la secuencia de proteína utilizando diversos ARN como intermediarios. Durante la transcripción genética, las secuencias de ADN son copiadas a ARN mediante una enzima llamada ARN polimerasa que sintetiza un ARN mensajero que mantiene la información de la secuencia del ADN. De esta manera, la transcripción del ADN también podría llamarse síntesis del ARN mensajero.
TRADUCCION :
La traducción es el segundo proceso de la síntesis proteica (parte del proceso general de la expresión génica). La traducción ocurre tanto en el citoplasma, donde se encuentran los ribosomas, como también en el retículo endoplasmático rugoso (RER). Los ribosomas están formados por una subunidad pequeña y una grande que rodean al ARNm. En la traducción, el ARN mensajero se decodifica para producir un polipéptido específico de acuerdo con las reglas especificadas por el código genético. Es el proceso que convierte una secuencia de ARNm en una cadena de aminoácidos para formar una proteína. Es necesario que la traducción venga precedida de un proceso de transcripción. El proceso de traducción tiene cuatro fases: activación, iniciación, elongación y terminación (entre todos describen el crecimiento de la cadena de aminoácidos, o polipéptido, que es el producto de la traducción).
En la activación, el aminoácido (AA) correcto se une al ARN de transferencia (ARNt) correcto. Aunque técnicamente esto no es un paso de la traducción, es necesario para que se produzca la traducción. El AA se une por su grupo carboxilo con el OH 3' del ARNt mediante un enlace de tipo éster. Cuando el ARNt está enlazado con un aminoácido, se dice que está "cargado". La iniciación supone que la subunidad pequeña del ribosoma se enlaza con el extremo 5' del ARNm con la ayuda de factores de iniciación (FI), otras proteínas que asisten el proceso. La elongación ocurre cuando el siguiente aminoacil-ARNt (el ARNt cargado) de la secuencia se enlaza con el ribosoma además de con un GTP y un factor de elongación. La terminación del polipéptido sucede cuando la zona A del ribosoma se encuentra con un codón de parada (sin sentido), que son el UAA, UAG o UGA. Cuando esto sucede, ningún ARNt puede reconocerlo, pero el factor de liberación puede reconocer los codones sin sentido y provoca la liberación de la cadena polipeptídica. La capacidad de desactivar o inhibir la traducción de la biosíntesis de proteínas se utiliza en antibióticos como la anisomicina, la cicloheximida, el cloranfenicol y la tetraciclina.
Yuri solis 2012-1313
ResponderEliminarPCR
El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato.
Desnaturalización Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación, Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada. La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la alcanza su máxima actividad. Estos últimos tienen tendencia a volver a unirse de modo espontáneo, ya que los extremos se pueden unir a otros extremos coincidentes que pueda haber en la cercanía (Apareamiento de Watson & Crick).
Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario.
La tecnica de Western blot y por lo tanto sigue los mismos paso solo se utiliza el western blot como una norma.
S outhern Blot para adn y proteina
El Southern Blot es una técnica que permite la identificación de secuencias específicas de DNA,mediante el uso de electroforesis en gel y de hibridación utilizando sondas especificas.Para analizar una muestra de DNA cromosomal ésta debe ser previamente fragmentada, por ejemploutilizando sonicación ó enzimas de restricción.Los fragmentos obtenidos se separan, de mayor a menor tamaño mediante una electroforesis en gel deagarosa. Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de DNA se traspasan aun filtro de nitrocelulosa ó a una membrana de nylon.A continuación, el filtro se incuba con una sonda marcada, específica para la secuencia que se deseaidentificar. Esta sonda es una secuencia de ácido nucleico, DNAds ó RNAm, que reconocerá a lasecuencia de DNA inmovilizada en el filtro, a través del reconocimiento de secuencias complementariasde ácidos nucleicos.El traspaso de las muestras desde el gel al filtro es una etapa esencial, porque el reconocimiento debases complementarias entre sonda y secuencias de DNA en el gel es muy ineficiente.Cabe señalar que el nombre de la técnica Southern Blot deriva en parte del apellido Southern delinvestigador que la desarrolló y de la palabra en inglés Blot que significa traspasar.
KERY JOUSSETT SANCHEZ ALVAREZ 2012-0919
ResponderEliminarTIPOS DE ARN:
El ARN mensajero (ARNm) es el tipo de ARN que lleva la información del ADN a los ribosomas, el lugar de la síntesis de proteínas. La secuencia de nucleótidos del ARNm determina la secuencia de aminoácidos de la proteína.21 Por ello, el ARNm es denominado ARN codificante.
No obstante, muchos ARN no codifican proteínas, y reciben el nombre de ARN no codificantes; se originan a partir de genes propios (genes ARN), o son los intrones rechazados durante el proceso de splicing. Son ARN no codificantes el ARN de transferencia (ARNt) y el ARN ribosómico (ARNr), que son elementos fundamentales en el proceso de traducción, y diversos tipos de ARN reguladores.22
Ciertos ARN no codificantes, denominados ribozimas, son capaces de catalizar reacciones químicas como cortar y unir otras moléculas de ARN,23 o formar enlaces peptídicos entre aminoácidos en el ribosoma durante la síntesis de proteínas.24
ARN implicados en la síntesis de proteínas.
Ribosoma 50S mostrando el ARNr (amarillo), las proteínas (azul) y el centro activo, la adenina 2486 (rojo).
ARN mensajero
El ARN mensajero (ARNm o RNAm) lleva la información sobre la secuencia de aminoácidos de la proteína desde el ADN, lugar en que está inscrita, hasta el ribosoma, lugar en que se sintetizan las proteínas de la célula. Es, por tanto, una molécula intermediaria entre el ADN y la proteína y apelativo de "mensajero" es del todo descriptivo. En eucariotas, el ARNm se sintetiza en el nucleoplasma del núcleo celular y donde es procesado antes de acceder al citosol, donde se hallan los ribosomas, a través de los poros de la envoltura nuclear.
ARN de transferencia[editar · editar código]
Los ARN de transferencia (ARNt o tRNA) son cortos polímeros de unos 80 nucleótidos que transfiere un aminoácido específico al polipéptido en crecimiento; se unen a lugares específicos del ribosoma durante la traducción. Tienen un sitio específico para la fijación del aminoácido (extremo 3') y un anticodón formado por un triplete de nucleótidos que se une al codón complementario del ARNm mediante puentes de hidrógeno.22
ARN ribosómico o ribosomal[editar · editar código]
El ARN ribosomico o ribosomal (ARNr o RNAr) se halla combinado con proteínas para formar los ribosomas, donde representa unas 2/3 partes de los mismos. En procariotas, la subunidad mayor del ribosoma contiene dos moléculas de ARNr y la subunidad menor, una. En los eucariotas, la subunidad mayor contiene tres moléculas de ARNr y la menor, una. En ambos casos, sobre el armazón constituido por los ARNr se asocian proteínas específicas. El ARNr es muy abundante y representa el 80% del ARN hallado en el citoplasma de las células eucariotas.25 Los ARN ribosómicos son el componente catalítico de los ribosomas; se encargan de crear los enlaces peptídicos entre los aminoácidos del polipéptido en formación durante la síntesis de proteínas; actúan, pues, como ribozimas.
ARN reguladores[editar · editar código]
Muchos tipos de ARN regulan la expresión génica gracias a que son complementarios de regiones específicas del ARNm o de genes del ADN.
ARN de interferencia[editar · editar código]
Los ARN interferentes (ARNi o iRNA) son moléculas de ARN que suprimen la expresión de genes específicos mediante mecanismos conocidos globalmente como ribointerferencia o interferencia por ARN. Los ARN interferentes son moléculas pequeñas (de 20 a 25 nucléotidos) que se generan por fragmentación de precursores más largos. Se pueden clasificar en tres grandes grupos:26
KERY JOUSSETT SANCHEZ ALVAREZ 2012-09019
ResponderEliminarARN
El ácido ribonucleico (ARN o RNA) es un ácido nucleico formado por una cadena de ribonucleótidos. Está presente tanto en las células procariotas como en las eucariotas, y es el único material genético de ciertos virus (virus ARN). El ARN celular es lineal y de hebra sencilla, pero en el genoma de algunos virus es de doble hebra.
En los organismos celulares desempeña diversas funciones. Es la molécula que dirige las etapas intermedias de la síntesis proteica; el ADN no puede actuar solo, y se vale del ARN para transferir esta información vital durante la síntesis de proteínas (producción de las proteínas que necesita la célula para sus actividades y su desarrollo). Varios tipos de ARN regulan la expresión génica, mientras que otros tienen actividad catalítica. El ARN es, pues, mucho más versátil que el ADN.
ADN
El ácido desoxirribonucleico, abreviado como ADN, es un ácido nucleico que contiene instrucciones genéticas usadas en el desarrollo y funcionamiento de todos los organismos vivos conocidos y algunos virus, y es responsable de su transmisión hereditaria. El papel principal de la molécula de ADN es el almacenamiento a largo plazo de información. Muchas veces, el ADN es comparado con un plano o una receta, o un código, ya que contiene las instrucciones necesarias para construir otros componentes de las células, como las proteínas y las moléculas de ARN. Los segmentos de ADN que llevan esta información genética son llamados genes, pero las otras secuencias de ADN tienen propósitos estructurales o toman parte en la regulación del uso de esta información genética.
Desde el punto de vista químico, el ADN es un polímero de nucleótidos, es decir, un polinucleótido. Un polímero es un compuesto formado por muchas unidades simples conectadas entre sí, como si fuera un largo tren formado por vagones. En el ADN, cada vagón es un nucleótido, y cada nucleótido, a su vez, está formado por un azúcar (la desoxirribosa), una base nitrogenada (que puede ser adenina→A, timina→T, citosina→C o guanina→G) y un grupo fosfato que actúa como enganche de cada vagón con el siguiente. Lo que distingue a un vagón (nucleótido) de otro es, entonces, la base nitrogenada, y por ello la secuencia del ADN se especifica nombrando sólo la secuencia de sus bases. La disposición secuencial de estas cuatro bases a lo largo de la cadena (el ordenamiento de los cuatro tipos de vagones a lo largo de todo el tren) es la que codifica la información genética: por ejemplo, una secuencia de ADN puede ser ATGCTAGATCGC... En los organismos vivos, el ADN se presenta como una doble cadena de nucleótidos, en la que las dos hebras están unidas entre sí por unas conexiones denominadas puentes de hidrógeno.
KERY JOUSSETT SANCHEZ ALVAREZ 2012-0919
ResponderEliminarCOMPUESTOS ORGANICOS
Compuesto orgánico o molécula orgánica es una sustancia química que contiene carbono, formando enlaces carbono-carbono y carbono-hidrógeno. En muchos casos contienen oxígeno, nitrógeno, azufre, fósforo, boro, halógenos y otros elementos menos frecuentes en su estado natural. Estos compuestos se denominan moléculas orgánicas. Algunos compuestos del carbono, carburos, los carbonatos y los óxidos de carbono, no son moléculas orgánicas. La principal característica de estas sustancias es que arden y pueden ser quemadas (son compuestos combustibles). La mayoría de los compuestos orgánicos se producen de forma artificial mediante síntesis química aunque algunos todavía se extraen de fuentes naturales.
Las moléculas orgánicas pueden ser de dos tipos:
Moléculas orgánicas naturales: son las sintetizadas por los seres vivos, y se llaman biomoléculas, las cuales son estudiadas por la bioquímica y las derivadas del petróleo como los hidrocarburos.
Moléculas orgánicas artificiales: son sustancias que no existen en la naturaleza y han sido fabricadas o sintetizadas por el hombre, por ejemplo los plásticos.
La línea que divide las moléculas orgánicas de las inorgánicas ha originado polémicas e históricamente ha sido arbitraria, pero generalmente, los compuestos orgánicos tienen carbono con enlaces de hidrógeno, y los compuestos inorgánicos, no. Así el ácido carbónico es inorgánico, mientras que el ácido fórmico, el primer ácido carboxilico, es orgánico. El anhídrido carbónico y el monóxido de carbono, son compuestos inorgánicos. Por lo tanto, todas las moléculas orgánicas contienen carbono, pero no todas las moléculas que contienen carbono son moléculas orgánicas.
KERY JOUSSETT SANCHEZ ALVAREZ 2012-0919
ResponderEliminarCOMPUESTOS INORGANICOS
Se denomina compuesto químico inorgánico a todos aquellos compuestos que están formados por distintos elementos, pero en los que su componente principal no siempre es el carbono, siendo el agua el más abundante. En los compuestos inorgánicos se podría decir que participan casi la totalidad de elementos conocidos. como el oro,plata .
Mientras que un compuesto orgánico se forma de manera natural tanto en animales como en vegetales, uno inorgánico se forma de manera ordinaria por la acción de varios fenómenos físicos y químicos: electrólisis, fusión, etc. También podrían considerarse agentes de la creación de estas sustancias a la energía solar, el agua, el oxígeno.
Los enlaces que forman los compuestos inorgánicos suelen ser iónicos o covalentes.
Ejemplos de compuestos inorgánicos:
Cada molécula de cloruro de sodio (NaCl) está compuesta por un átomo de sodio y otro de cloro.
Cada molécula de agua (H2O) está compuesta por dos átomos de hidrógeno y uno de oxígeno.
Cada molécula de amoníaco (NH3) está compuesta por un átomo de nitrógeno y tres de hidrógeno.
El anhídrido carbónico se encuentra en la atmósfera en estado gaseoso y los seres vivos aerobios lo liberan hacia ella al realizar la respiración. Su fórmula química, CO2, indica que cada molécula de este compuesto está formada por un átomo de carbono y dos de oxígeno. El CO2 es utilizado por algunos seres vivos autótrofos como las plantas en el proceso de fotosíntesis para fabricar glucosa. Aunque el CO2 contiene carbono, no se considera como un compuesto orgánico porque no contiene hidrógeno.
KERY JOUSSETT SANCHEZ ALVAREZ 2012-0919
ResponderEliminarACIDOS NUCLEICOS
El descubrimiento de los ácidos nucleicos se debe a Friedrich Miescher, quien en el año 1869 aisló de los núcleos de las células una sustancia ácida a la que llamó nucleína,1 nombre que posteriormente se cambió a ácido nucleico. Posteriormente, en 1953, James Watson y Francis Crick descubrieron la estructura del ADN, empleando la técnica de difracción de rayos X.
Los ácidos nucleicos son grandes polímeros formados por la repetición de monómeros denominados nucleótidos, unidos mediante enlaces fosfodiéster. Se forman, así, largas cadenas; algunas moléculas de ácidos nucleicos llegan a alcanzar tamaños gigantescos, con millones de nucleótidos encadenados. Los ácidos nucleicos almacenan la información genética de los organismos vivos y son los responsables de la transmisión hereditaria. Existen dos tipos básicos, el ADN y el ARN.
Formula de los acidos nucleicos
X+Y- xy X y + pn =
TIPOS
Existen dos tipos de ácidos nucleicos: ADN (ácido desoxirribonucleico) y ARN (ácido ribonucleico), que se diferencian:
por el glúcido (la pentosa es diferente en cada uno; ribosa en el ARN y desoxirribosa en el ADN);
por las bases nitrogenadas: adenina, guanina, citosina y timina, en el ADN; adenina, guanina, citosina y uracilo, en el ARN;
en la inmensa mayoría de organismos, el ADN es bicatenario (dos cadenas unidas formando una doble hélice), mientras que el ARN es monocatenario (una sola cadena), aunque puede presentarse en forma extendida, como el ARNm, o en forma plegada, como el ARNt y el ARNr;
en la masa molecular: la del ADN es generalmente mayor que la del ARN.
KERY JOUSSETT SANCHEZ ALVAREZ 2012-0919
ResponderEliminarLas proteínas son moléculas formadas por cadenas lineales de aminoácidos. El término proteína proviene de la palabra francesa protéine y ésta del griego πρωτεῖος (proteios), que significa 'prominente, de primera calidad'.1
Por sus propiedades físico-químicas, las proteínas se pueden clasificar en proteínas simples (holoproteidos), que por hidrólisis dan solo aminoácidos o sus derivados; proteínas conjugadas (heteroproteidos), que por hidrólisis dan aminoácidos acompañados de sustancias diversas, y proteínas derivadas, sustancias formadas por desnaturalización y desdoblamiento de las anteriores. Las proteínas son necesarias para la vida, sobre todo por su función plástica (constituyen el 80% del protoplasma deshidratado de toda célula), pero también por sus funciones biorreguladoras (forman parte de las enzimas) y de defensa (los anticuerpos son proteínas).2
Las proteínas desempeñan un papel fundamental para la vida y son las biomoléculas más versátiles y diversas. Son imprescindibles para el crecimiento del organismo y realizan una enorme cantidad de funciones diferentes, entre las que destacan:
Estructural. Esta es la función más importante de una molecula
(Ej: colágeno),
Inmunológica (anticuerpos),
Enzimática (Ej: sacarasa y pepsina),
Contráctil (actina y miosina).
Homeostática: colaboran en el mantenimiento del pH (ya que actúan como un tampón químico),
Transducción de señales (Ej: rodopsina)
Protectora o defensiva (Ej: trombina y fibrinógeno)
Las proteínas están formadas por aminoácidos los cuales a su vez están formados por enlaces peptídicos para formar esfingosinas.
Las proteínas de todos los seres vivos están determinadas mayoritariamente por su genética (con excepción de algunos péptidos antimicrobianos de síntesis no ribosomal), es decir, la información genética determina en gran medida qué proteínas tiene una célula, un tejido y un organismo.
Las proteínas se sintetizan dependiendo de cómo se encuentren regulados los genes que las codifican. Por lo tanto, son susceptibles a señales o factores externos. El conjunto de las proteínas expresadas en una circunstancia determinada es denominado proteoma.
marcel lus arredondo 2012-1215
ResponderEliminarREACCIÓN EN CADENA DE LA POLIMERASA (PCR)
Este método se basa en las características de la estructura química del DNA y su replicación. El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato. Específicamente el hidroxilo 3 del azúcar de un desoxirribonucleótido se une al hidroxilo 5 del siguiente azúcar a través del puente fosfodiester. Para duplicarse, el ADN se separa en sus dos cadenas complementarias; así, cada una de ellas sirve de molde. Al final del proceso se obtendrán 2 moléculas de ADN, cada una de ellas formada por una cadena original y una cadena complementaria que ha sido sintetizada
PCR
El ADN es un polímero formado por 2 cadenas complementarias, constituido por unidades de desoxirribonucleótidos de 4 bases nitrogenadas adenina, guanina, citosina y timina unidas al azúcar desoxirribosa. La parte que no varía en la molécula del ADN consiste en desoxirribosa unidas por grupos fosfato.
Desnaturalización Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación, Hibridación: Luego que ocurre la desnaturalización, se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, Extensión: En esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada
ACIDOS NUCLEICOS
Los ácidos nucleicos son grandes polímeros formados por la repetición de monómeros denominados nucleótidos, unidos mediante enlaces fosfodiéster. Se forman, así, largas cadenas; algunas moléculas de ácidos nucleicos llegan a alcanzar tamaños gigantescos, con millones de nucleótidos encadenados. Los ácidos nucleicos almacenan la información genética de los organismos vivos y son los responsables de la transmisión hereditaria. Existen dos tipos básicos, el ADN y el ARN.
El ADN MITOCONDRIAL
Es un fragmento de ácido nucleico encontrado dentro de las mitocondrias. Las mitocondrias son pequeños organelos o estructuras celulares que se encuentran dentro de las células eucarióticas (generalmente las células animales). Estos organelos se usan básicamente para la producción y generación de energía para que la célula lleve a cabo sus funciones. Las plantas tienen cloroplastos. Las células animales tienen mitocondrias. Y cada una de estas mitocondrias tiene su propio genoma. Su propia molécula circular de ADN. Y eso es el ADN mitocondrial, porque en realidad es diferente del ADN que se encuentra en el núcleo de las células. No se encuentra dentro del núcleo. El número de genes en el ADN mitocondrial es de 37, frente a los 20.000 - 25.000 genes del ADN cromosómico nuclear humanos.Codifica dos ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa. El ADN mitocondrial está en replicación constante, independientemente del ciclo y del tipo celular.
ELISA DIRECTA E INDIRECTA
Un ensayo de inmunoabsorción ligado a enzimas (ELISA) implica la reacción de un complejo anticuerpo-enzima con un antígeno o anticuerpo que se mantiene inmóvil sobre una superficie sólida. En la incubación de la enzima conjugada con un sustrato adecuado, un producto es formado. La formación de un producto ayuda a detectar la presencia del antígeno o anticuerpo y su cantidad proporciona una medida de la cantidad de antígeno-anticuerpo de la reacción que se produjo. La prueba de ELISA se puede realizar de dos formas: directa e indirecta.
Ginsely Jiménez Tavárez 2012-1217
ResponderEliminarADN Mitocondrial:También llamado cromosoma mitocondrial, es una molécula circular de DNA de un tamaño de 16569 pares de bases (bp) (8000 veces menor que el cromosoma medio). Este tamaño de 16569 bp corresponde al primer ADN secuenciado (secuencia Cambridge) aunque existen otras variantes con un número de pares de bases que oscila entre 16559 y 16570. En cada mitocondria existen varias copias de este ADN, de modo que el número de cromosomas mitocondriales en cada célula puede ser de varios miles. Cuatro o cinco cromosomas mitocondriales se agrupan formando los llamados nucleoides. El ADN mitocondrial es muy parecido al del los cromosomas bacterianos, estando formado por dos cadenas complementarias, cada una con 16569 pares de bases, pero con un peso molecular diferente: la cadena pesada H (peso molecular, 5.168.726 daltons) contiene muchas más G que la cadena ligera L (peso molecular, 5.060.609 daltons). La mayor parte de la cadena H constituye el molde para la transcripción de la mayor parte de los genes, mientras que la cadena L es la cadena codificadora.El genoma mitocondrial contiene un total de 37 genes de los cuales 13 genes que codifican para ARNs mensajeros, y por lo tanto para 13 proteínas, 22 genes que codifican para 22 tARNs (ARNs de transferencia, que se representan simbólicamente como hojas de trebol) y 2 genes que codifican para dos rRNAs mitocondriales (RNAs ribosómicos). En la práctica, cuando se secuencia el ADN miocrondrial se unna persona en particular, se observan un cierto número de variaciones que son simplemente polimorfismos no tienen ninguna consecuencia clínica. De hecho, una región del ADN-mitocondrial llamada región de control, es tan polimórfica que se utiliza en medicina forense para identificar al sujeto.Además de las proteínas que la mitocondria puede sintetizar por si misma, necesita importar algunas otras sintetizadas en el núcleo. De igual forma, los lípidos que forman las membranas externa e interna de la mitocondria son importadas.Las mutaciones de algunos de los genes mitocondriales ocasionan enfermedades en el hombre. Se conocen las siguientes mutaciones:MELAS: (miopatía mitocondrial con encefalopatía, acidosis láctica y episodios similares al ictus). Se debe a una disfunción el complejo I de la cadena respiratoria mitocondrial debida a un cambio de bases en el par 3243 de la cadena pesadaMERRF: (epilepsia mioclónica, fibras rojas deshilachadas): se debe sobre todo a una mutación del gen que codifica el t-ARN de la lisina por un cambio de bases en la posición 8344 de la cadena pesada. Este cambio produce una disfunción del complejo V de la cadena respiratoria.
lucisol cano 2012-1254
ResponderEliminarARN
El ácido ribonucleico (ARN o RNA) es un ácido nucleico formado por una cadena de ribonucleótidos. Está presente tanto en las células procariotas como en las eucariotas, y es el único material genético de ciertos virus (virus ARN). El ARN celular es lineal y de hebra sencilla, pero en el genoma de algunos virus es de doble hebra.
En los organismos celulares desempeña diversas funciones. Es la molécula que dirige las etapas intermedias de la síntesis proteica; el ADN no puede actuar solo, y se vale del ARN para transferir esta información vital durante la síntesis de proteínas (producción de las proteínas que necesita la célula para sus actividades y su desarrollo). Varios tipos de ARN regulan la expresión génica, mientras que otros tienen actividad catalítica. El ARN es, pues, mucho más versátil que el ADN.
ADN
El ácido desoxirribonucleico, abreviado como ADN, es un ácido nucleico que contiene instrucciones genéticas usadas en el desarrollo y funcionamiento de todos los organismos vivos conocidos y algunos virus, y es responsable de su transmisión hereditaria. El papel principal de la molécula de ADN es el almacenamiento a largo plazo de información. Muchas veces, el ADN es comparado con un plano o una receta, o un código, ya que contiene las instrucciones necesarias para construir otros componentes de las células, como las proteínas y las moléculas de ARN. Los segmentos de ADN que llevan esta información genética son llamados genes, pero las otras secuencias de ADN tienen propósitos estructurales o toman parte en la regulación del uso de esta información genética.
Lucisol Cano 2012-1254
ResponderEliminarCOMPUESTOS INORGANICOS
Se denomina compuesto químico inorgánico a todos aquellos compuestos que están formados por distintos elementos, pero en los que su componente principal no siempre es el carbono, siendo el agua el más abundante. En los compuestos inorgánicos se podría decir que participan casi la totalidad de elementos conocidos. como el oro,plata .
Mientras que un compuesto orgánico se forma de manera natural tanto en animales como en vegetales, uno inorgánico se forma de manera ordinaria por la acción de varios fenómenos físicos y químicos: electrólisis, fusión, etc. También podrían considerarse agentes de la creación de estas sustancias a la energía solar, el agua, el oxígeno.
Los enlaces que forman los compuestos inorgánicos suelen ser iónicos o covalentes.
Ejemplos de compuestos inorgánicos:
Cada molécula de cloruro de sodio (NaCl) está compuesta por un átomo de sodio y otro de cloro.
Cada molécula de agua (H2O) está compuesta por dos átomos de hidrógeno y uno de oxígeno.
Cada molécula de amoníaco (NH3) está compuesta por un átomo de nitrógeno y tres de hidrógeno.
Lucisol Cano 2012-1254 Tecnicas Blotting
ResponderEliminarEl primer paso de todo Western Blot es la separación de las macromoléculas mediante geles de electroforesis; después de la misma, las macromoléculas ya separadas en función de su diferente peso molecular se transfieren a una segunda matriz, generalmente una membrana de nitrocelulosa o PVDF. Posteriormente, se bloquea la membrana para evitar la unión inespecífica a su superficie de los anticuerpos que se van a utilizar para la detección de la proteína de interés. En el siguiente paso, se une a dicha proteína transferida un anticuerpo especifico marcado con una enzima y, finalmente, se añade un sustrato apropiado para dicha enzima con lo que se produce un producto detectable como, por ejemplo, un precipitado cromogénico o fluorogénico en la membrana. En la actualidad, los métodos más sensibles emplean sustratos quimioluminiscentes que cuando se combinan con la enzima correspondiente, producen luz como producto final, que puede detectarse mediante una película o una cámara CCD. En todos los casos y sea cual sea el sustrato que se utilice, la intensidad de la señal se correlaciona con la cantidad del antígeno en la superficie de la membrana.
TEMA10: ELECTROFORESIS DE PROTEÍNAS EN SUERO
ResponderEliminarEste examen mide los tipos de proteína en la parte líquida (suero) de una muestra de sangre.
Ver también:
Forma en que se realiza el examen
Se necesita una muestra de sangre. Para obtener información con respecto a dar una muestra de sangre de una vena, ver el artículo: venopunción
La electroforesis es una técnica de laboratorio, en la cual el suero sanguíneo (la parte líquida de la sangre sin células) se coloca en un papel especialmente tratado y luego se expone a una corriente eléctrica. Las proteínas en el suero se mueven en el papel para formar bandas que muestran la proporción de cada fracción de proteína. Una fracción puede contener varios tipos diferentes de proteínas.
Las proteínas individuales, con excepción de la albúmina, generalmente no se miden; sin embargo, sí se miden las fracciones o grupos de proteínas. Los niveles de las fracciones de proteínas se pueden calcular midiendo la proteína sérica total y luego multiplicando eso por el porcentaje relativo de cada fracción de proteína.
La electroforesis de lipoproteínas es un tipo de electroforesis de proteínas que determina la cantidad de proteínas compuestas de proteína y grasa, llamadas lipoproteínas (como el colesterol LDL).
Lucisol Cano 2012-1254
ResponderEliminarTema 3 - ELISA…
El ensayo inmunoabsorbente ligado a enzimas (ELISA) utiliza como sus siglas lo indican una enzima como marcador para mediar la formación de complejos antígeno-anticuerpo.
Existen diversas variaciones al método de ELISA para detectar y cuantificar ligandos de alto peso molecular (>30000 daltons), el marcador enzimático que se emplea en estos análisis se conjuga con un ligando, que puede ser un antígeno, un anticuerpos específico para el antígeno de interés o un anticuerpo para el anticuerpo primario. Casi todas las pruebas ELISA son ensayos en fase sólida en los cuales se adsorbe un antígeno o un anticuerpo sobre un soporte sólido. Algunos de los protocolos se basan en reacciones de enlace competitivo y otras en reacciones de enlace no competitivo, pero en todas las pruebas ELISA se requiere de un paso de separación para eliminar el conjugado enzimático libre antes de proceder a determinar la cantidad de conjugado enzimático enlazado. Para lo cual se añade sustrato enzimático y se mide la reacción catalítica entre la enzima y el sustrato. Por sus características catalíticas las enzimas son marcadores muy sensibles y versátiles. Una sola proteína enzimática puede transformar en algunos minutos gran número de moléculas de sustrato en una cantidad igualmente abundante de producto final, produciendo un cambio de color amplificado y que se detecta con facilidad.En el método ELISA de inhibición el cambio de color se reduce, porque la actividad enzimática se inhibe cuando el anticuerpo se enlaza al conjugado enzimático.
Lucisol Cano 2012-1254
EliminarTEMA 7 :El Western blot, o inmunoblot, es una técnica analítica usada para detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis en gel se separan las proteínas atendiendo al criterio que se desee: peso molecular, estructura, hidrofobicidad, etc. Hay casi tantas posibilidades como tipos de electroforesis existen. Luego son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella.1 2 Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas.
Esta técnica es hoy en día imprescindible en varios campos de la biología, como la biología molecular, la bioquímica, la biotecnología o la inmunología. Existe toda una industria especializada en la venta de anticuerpos (monoclonales y policlonales) contra decenas de miles de proteínas diferentes.3 4
ALESHA LOW 2012-1318
ResponderEliminarMANEJO DE MUESTRAS Y REACTIVOS.
El laboratorio de biología celular y molecular tiene una serie de condiciones
especiales relacionadas con el manejo de reactivos, elementos y equipos que
pueden influir en la salud del personal así como repercutir en el medio ambiente.
Por ende, es importante conocer las normas establecidas, familiarizarse con el uso
de cada uno de los materiales de trabajo para evitar cometer errores o sufrir
accidentes durante su manipulación.
El personal del laboratorio, por lo tanto, está expuesto a una serie de riesgos
ocupacionales por agentes físicos, químicos y biológicos, los cuales pueden
originar alteraciones en la salud, generalmente prevenibles si se aplican las
normas de bioseguridad, que incluyen el manejo correcto de material infeccioso,
así como el uso de elementos protectores, y el manejo adecuado de desechos
biológicos y reactivos químicos. Al manipular reactivos químicos, es necesario conocer y evaluar sus peligros y
determinar la forma apropiada para su manejo seguro. Esta información se obtiene
de las etiquetas, hojas de seguridad suministradas por el fabricante u otras
fuentes.
Las etiquetas del fabricante incluyen el nombre del producto, el pictograma de
seguridad, las frases de riesgo entre otras. El etiquetado del puesto de trabajo se
emplea cuando el producto es transferido a otro contenedor diferente al original.
Allí debe contener el nombre del producto, del fabricante, frases de riesgo y
consejos de prudencia.
Las hojas de seguridad informan sobre las propiedades físicas, químicas y
toxicológicas de una sustancia y los procedimientos recomendados para una
manipulación segura, uso de equipos para protección, primeros auxilios,
tratamiento de desechos, derrames y otros.
La clasificación de las sustancias químicas depende de su peligrosidad, de lo cual
se establece que existen:
• Tóxicas
• Muy tóxicas
• Corrosivas
• Irritantes
• Inflamables
• Extremadamente inflamables
Los riegos biológicos se pueden identificar según: el tipo de material biológico que
se manipula, los procedimientos de rutina realizados en el laboratorio, el manejo
de desechos biológicos, el aseo y desinfección de las áreas de trabajo, la
disponibilidad de implementos de protección personal, los protocolos de
reconocimiento, las enfermedades relacionadas con el trabajo, los accidentes e
incidentes más comunes como pinchazos, cortaduras e inhalaciones, las
enfermedades ocupacionales adquiridas en el laboratorio.
ALESHA LOW 2012-1318
ResponderEliminarREACCION EN CADENA DE LA POLIMERASA.
Durante las décadas de 1970-1980, la manera más práctica de hacer múltiples copias de una secuencia particular de ADN era introduciendo una molécula de ADN recombinante (un plásmido más un gen) en una célula huésped. Esto podía resultar un poco engorroso, ya que muchas veces se disponía de una cantidad muy pequeña de moléculas de ADN para realizar pruebas.
A mediados de la década de los 80, la invención de una técnica capaz de generar centenares de miles de copias de una secuencia sin la necesidad de clonarla en ningún tipo de vector resolvió este problema.
Kary Mullis, un bioquímico americano que trabajaba sintetizando ADN, resolvió este problema con la invención de la reacción en cadena de la polimerasa.
Esta se basa en la separación de ambas hebras de ADN, la posterior unión de pequeños fragmentos (denominados primers) y la extensión de estos fragmentos usando de molde el ADN de la muestra. Así se obtienen dos copias idénticas por cada molécula en cada ciclo de reacción. Para llevar a cabo esta reacción se requiere de una enzima polimerasa que sea capaz de soportar la alta temperatura del proceso de desnaturalización. Esta enzima es una ADN-polimerasa especial obtenida de una bacteria termófila (la Thermus aquaticus), resistente a altas temperaturas.
La reacción en cadena de la polimerasa permite amplificar la muestra de manera espectacular y su sensibilidad es tal que alcanza con una molécula para que la reacción genere una infinidad de copias. Esto la convierte en una importante, si no imprescindible, herramienta para el análisis de filiación o de criminalística forense, ya que con sólo una pequeñísima muestra se pueden realizar diferentes estudios comparativos que nos permiten conocer el dueño de un “rastro” genético en particular.
También se usa para el desarrollo de nuevas estrategias de diagnóstico médico, como la detección de virus o de mutaciones que provocan enfermedades genéticas, a partir de una muestra de ADN tan pequeña como un cabello o una gota de sangre.
Otra aplicación de la técnica de la reacción en cadena de la polimerasa es la transcripción inversa de ARNm. Mediante el uso de una enzima de origen viral denominada transcriptasa reversa puede usarse como molde una molécula de ARNm, transcribirla a una secuencia de ADN, y luego ser amplificada como cualquier otra secuencia por la reacción en cadena de la polimerasa, con lo que se obtiene una gran cantidad de ADN a partir de unas pocas moléculas de ARNm. Esta técnica es muy empleada para el estudio de expresión génica, así como para la producción de proteínas en diferentes organismos.
ALESHA LOW 2012-1318
ResponderEliminarPCR
ETAPAS DE LA PCR
Desnaturalización: Para que pueda iniciarse la reacción es preciso que las moléculas de ADN molde se encuentren en forma de cadena simple. Esto se consigue calentando a temperatura de 90 a 95ºC para que produzca la rotura de los enlaces puente de hidrogeno intercatenarios y la separación de ambas cadenas, para asegurar la completa separación de la doble cadena del ADN esta temperatura debe mantenerse unos minutos. Si el ADN solo se desnaturaliza parcialmente éste tenderá a renaturalizarse muy rápidamente dificultando con esto el proceso de hibridación.
Hibridación: Una vez que el ADN está desnaturalizado se disminuye la temperatura de la reacción hasta un rango comprendido entre los 40 y los 60ºC para que se pueda producir la hibridación específica de los cebadores a las secuencias flanqueantes del fragmento que se desea amplificar, la temperatura a la que se realiza esta etapa debe establecerse para cada reacción en función de la longitud de los cebadores y su secuencia (Temperaturas inferiores a la óptima nos producirán hibridaciones inespecíficas de los cebadores y temperaturas superiores nos dificultarán la eficiencia de la misma.
Extensión: Durante esta etapa la ADN polimerasa termoresistente incorpora nucleótidos en el extremo 3´ del cebador utilizado como molde la cadena de ADN previamente desnaturalizada.
La temperatura a la que se realiza esta etapa de la reacción suele ser de 72ºC ya que es la temperatura a la que la “Taq polimerasa” alcanza su máxima actividad. Normalmente una extensión de 20 segundos es suficiente para fragmentos menores de 500 pares de bases, y 40 segundos para fragmentos por encima de 1.2Kb.Al finalizar cada uno de los ciclos el número de copias obtenidas se duplica y después de 20 ciclos ya tenemos aproximadamente 1 millón de copias de cada una de las moléculas molde iniciales ADN.
ALESHA LOW 2012-1318
ResponderEliminarENZIMAS DE RESTRICCION (BRENDA)
Las enzimas de restricción son nucleasas que cortan ADN de doble cadena cuando reconocen un patrón de secuencia específico. Generan fragmentos de ADN conocidos como fragmentos de restricción. Las enzimas de restricción son herramientas imprescindibles en biología molecular, ingeniería genética y biotecnología.
Las enzimas de restricción son nucleasas que cortan el ADN cuando reconocen un patrón de secuencia específico.
Se descubrieron en los años 60 y su uso en biología molecular empezó en los 70 permitiendo el desarrollo de las tecnologías de ADN recombinante. Las enzimas de restricción tienen actividad endonucleasa y cortan enlaces fosfodiéster en las dos cadenas del ADN. Reconocen ciertas secuencias en el ADN. Las enzimas de restricción de tipo I y III reconocen una secuencia específica pero cortan a una distancia variable del sitio de reconocimiento (sitio de restricción). Las enzimas de restricción de tipo II reconocen una secuencia específica conocida como secuencia diana y siempre cortan entre los mismos nucleótidos. De ahí que generen siempre los mismos fragmentos de restricción. La secuencia diana es de tamaño variable, la mayoría de 4 y 6 nucleótidos, y con frecuencia es parcialmente palindrómica. Algunas enzimas de restricción cortan generando extremos llamados romos mientras que otras cortan generando extremos llamados cohesivos.
Las enzimas de restricción son una herramienta básica en Biología molecular: en laboratorios de PCR, creación de sondas para hibridación, laboratorios de secuenciación de ADN, ingeniería genética, procesos de clonación, manejo de genotecas y en muchas otras áreas de genómica.
ALESHA LOW 2012-1318
ResponderEliminarBRENDA
Las enzimas de restricción, también conocidas como endonucleasas, son enzimas que cortan los enlaces fosfodiester del material genético a partir de una secuencia que reconocen.
· Las mismas permiten cortar DNA de hebra doble, donde reconocen secuencias palindrómicas (secuencias que se leen igual en ambas direcciones).
· Son extraídas de organismos procarióticos (bacterias), donde actúan como un mecanismo de defensa, para degradar material genético extraño que entre en la célula. Las bacterias tienen la capacidad de metilar su DNA, lo cual sirve para distinguir entre el DNA extraño y el DNA propio. Las enzimas de restricción no pueden cortar DNA metilado, de este modo solo afectan el DNA extranjero y no el DNA bacterial.
EcoRI es una enzima de restricción producida por el microorganismo Escherichia coli que posee una diana de restricción en el ADN de cadena doble dependiente de una secuencia metilada (al estar la hebra metilada, no hay corte), palindrómica y asimétrica, sobre la cual su actividad catalítica hidrolasa genera extremos cohesivos. Es sin duda la enzima más conocida dentro del mundo académico. La forma correcta de nombrarla es "Eco erre 1". En adision es la mas utilizada en laboratorios de universidades para beneficiar al estudiante y que aprenda como manejar con enzimas de restricción.
ALESHA LOW 2012-1318
ResponderEliminarELISA DIRECTO (SANDWICH)
ELISA directo (ensayo ELISA simple de dos capas). Las placas ELISA se preparan recubriendo los pocillos con las soluciones en las que se sospecha se encuentra el antígeno. Se incuban con anticuerpos marcados. Indican la presencia de antígeno en la solución analizada. Es necesario incluir controles negativos que serán muestras del mismo tipo de las analizadas (sangre, orina,...) pero en las que se tenga la certeza de la ausencia del antígeno buscado. Asimismo se incluyen controles positivos (soluciones donde se encuentra el antígeno buscado).
ALESHA LOW 2012-1318
ResponderEliminarELISA DIRECTO SANDWICH
ELISA ''sándwich'' (Ensayo de captura de antígeno y detección mediante inmunocomplejos). Se trata de un ensayo muy empleado en el que se recubre el pocillo con un primer anticuerpo anti-antígeno. Después de lavar el exceso de anticuerpo se aplica la muestra problema en la que se encuentra el antígeno, que será retenido en el pocillo al ser reconocido por el primer anticuerpo. Después de un segundo lavado que elimina el material no retenido se aplica una solución con un segundo anticuerpo anti-antígeno marcado. Así pues cada molécula de antígeno estará unida a un anticuerpo en la base que lo retiene y un segundo anticuerpo, al menos, que lo marca. Este ensayo tiene una gran especificidad y sensibilidad debido a la amplificación de señal que permite el segundo anticuerpo.
ALESHA LOW 2012-1318
ResponderEliminarELISA INDIRECTO
ELISA indirecto. Las placas ELISA se preparan de la misma forma a la anterior. Los controles positivos y negativos son los mismos. El sistema de detección emplea dos anticuerpos: uno primario contra el antígeno y uno secundario marcado contra el primario. La detección tiene mayor sensibilidad por presentar una amplificación de señal debida a la unión de dos o más anticuerpos secundarios por cada primario. Es el ensayo más popular, como lo es la inmunofluorescencia indirecta, pues un mismo secundario marcado y un mismo sistema enzimático permite cuantificar una gran variedad de antígenos, por eso es un método más polivalente y barato, aunque se pierda algo de precisión por tener un eslabón más con respecto al método directo. La dilución de la solución que contiene el anticuerpo primario (por ejemplo: suero sanguíneo) es un factor muy importante a tener en cuenta para evitar la aparición de falsos negativos, ya que si la muestra está muy diluida no saldrá positiva si la titulación de anticuerpos es muy baja. Es decir, aunque los anticuerpos están presentes, la prueba no da positivo porque la concentración de anticuerpos específicos contra el antígeno que está pegado en el fondo del pocillo no es suficiente como para dar una señal detectable.
El ELISA indirecto es el método de elección para detectar la presencia de anticuerpos séricos contra el virus de la inmunodeficiencia humana (VIH), agente causante del síndrome de inmunodeficiencia adquirida (SIDA). Según esta técnica, proteínas recombinantes de la envoltura y el núcleo del VIH se absorben como antígenos en fase sólida a los pocillos. Las personas afectadas de VIH producen anticuerpos séricos contra epítopos en estas proteínas víricas. En general, el ELISA indirecto permite detectar anticuepos séricos contra VIH desde las primeras seis semanas de una infección.
ALESHA LOW 2012-1318
ResponderEliminarELISA INDIRECTO
En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA.
ALESHA LOW 2012-1318
ResponderEliminarSOUTHERN BLOT PARA DNA
Southern blot, hibridación Southern o, simplemente, Southern es un método de biología molecular que permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este ácido nucleico. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de separar los fragmentos de ADN de acuerdo a su longitud y, después, una transferencia a una membrana en la cual se efectúa la hibridación de la sonda. Su nombre procede del apellido de su inventor, un biólogo inglés llamado Edwin Southern.1. Extracción del ADN
Se puede extraer ADN de casi cualquier tejido humano. Las posibles fuentes de ADN en la escena de un delito incluyen sangre, semen, tejido de una víctima muerta, células del folículo capilar y saliva. El ADN extraído de las pruebas del delito ("indicios" o "vestigios" biológicos) se compara con el extraído de muestras de referencia, obtenidas de personas conocidas,habitualmente de la sangre.
2. Digestión del ADN con una endonucleasa de restricción
El ADN extraído de la muestra se trata con una endonucleasa de restricción,que es una enzima que corta el ADN bicatenario en donde tenga una secuencia característica. La enzima que se usa más frecuentemente para el análisis legal es HaeIII, que corta el ADN en la secuencia 5'-GGCC-3'.
3. Electroforesis en gel de agarosa
Tras la digestión del ADN, los fragmentos de ADN resultantes se separan según su tamaño mediante electroforesis en geles de agarosa. Durante la electroforesis, las moléculas de ADN, que poseen carga negativa, migran hacia el electrodo positivo. Al avanzar las moléculas de ADN, su velocidad de migración se ve reducida por la matriz del gel de agarosa. Las moléculas menores se mueven más deprisa a través de los poros del gel que las de mayor tamaño. Como resultado, se produce una separación continua de los fragmentos de ADN de acuerdo con su tamaño, de modo que los fragmentos más pequeños avanzan la mayor distancia con referencia al origen o punto de aplicación de la muestra.
4. Preparación de un ensayo de Southern ("Southern blot")
Tras la electroforesis, las moléculas de ADN separadas se desnaturalizan mientras permanecen en el gel de agarosa, impregnando éste con una disolución alcalina. Tras la neutralización de ésta, el DNA monocatenario resultante se transfiere a la superficie de una membrana de nailon,realizando así una copia o "calco" (la traducción más literal del inglés blot,conservando este sentido, es "secante"). Este proceso de desnaturalización y transferencia se conoce como método de Southern en recuerdo de quien lo inventó, Edward Southern. Al igual que la aplicación de un secante a un papel con la tinta húmeda transfiere una réplica de la imagen del papel al secante,el "calco" del ADN en el gel a la membrana de nailon conserva la distribución espacial de los fragmentos de ADN conseguida en el gel como resultado de la electroforesis.
ALESHA LOW 2012-1318
ResponderEliminarNORTHERN BLOT PARA DNA
Northern blot es una técnica de detección de moléculas de ácido ribonucleico (ARN) de una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN mensajero para un péptido dado en una extracción de ARN total). Para ello, se toma la mezcla de ARN y se somete a una electroforesis en gel a fin de separar los fragmentos en base a su tamaño. Tras esto, se transfiere el contenido del gel, ya resuelto, a una membrana cargada positivamente en la cual se efectúa la hibridación de una sonda molecular marcada radiactiva o químicamente.
El nombre de la técnica deriva de la propia de la detección de ácido desoxirribonucleico (ADN), denominada Southern blot en honor a su descubridor; de este modo, al desarrollarse la técnica equivalente para ARN se empleó el punto cardinal opuesto («northern», septentrional en inglés, frente al meridional «southern»). El northern blot fue desarrollado en 1977 por James Alwine, David Kemp y George Stark en la Universidad de Stanford.El procedimiento general comienza con la extracción del RNA total de una muestra de tejido homogenizado. El mRNA se puede aislar a través del uso de cromatografía para mantener solamente los ARN con colas de poli-A. Las muestras de ARN son entonces separadas por electroforesis en gel. Dada la fragilidad de los geles y la incapacidad de las sondas para penetrar en la matriz, las muestras de RNA, separadas por tamaño tras la electroforesis, serán tranferidas a una membrana de Nailon por medio de capilaridad o un sistema de transferencia al vacío.
Los sistemas de capilaridad iónica se utilizan pra transferir el ARN desde un gel de electroforesis a una membrana de Northern blot.
Una membrana de Nylon cargada positivamente es el soporte mas efectivo para usar en el northern blot ya que los ácidos nucleicos, que están cargados negativamente, tienen una alta afinidad por estas membranas. El buffer de transferencia usado para el ensayo suele contener formamida, dado que ésta consigue disminuir la temperatura de hibridación de la sonda, lo cual previene la degradación del ARN por las altas temperaturas. Una vez que el ARN ha sido transferido a la membrana, este es inmovilizado a través de enlaces covalentes formados con la membrana, lo cual se consigue por medio de luz ultravioleta o calor. Después del marcaje de la sonda, ésta se hibrida con el RNA en la membrana. Las condiciones experimentales que pueden afectar la eficiencia y especificidad de la hibridación están determinadas por las condiciones iónicas, de viscosidad, la presencia de dobles hebras de RNA, bases desemparejadas y composición de las mismas. Finalmente, la membrana debe de lavarse para asegurar que la sonda se ha unido de forma específica y para evitar ruido de fondo en las señales emitidas por la misma. Estas señales serán detectadas por Rayos X y pueden ser cuantificadas mediante técnicas de densitometría. Para crear controles y poder asegurarnos de que no se están mostrando genes que no nos interesen se puede realizar posteriormente la determinación por microarrays o RT-PCR.
ALESHA LOW 2012-1318
ResponderEliminarEASTERN BLOTT PARA EPITOPOS CARBOHIDRATOS
EASTERN BLOT es una técnica bioquímica utilizado para analizar proteínas modificaciones post traduccionales (PTM), tales como lípidos, phosphomoieties y glicoconjugados. Con mayor frecuencia se utiliza para detectar carbohidratos epítopos . Por lo tanto, este secante puede ser considerado una extensión de la técnica de bioquímica de transferencia Western . Varias técnicas han sido descritas por el término del Este secante, proteínas más uso borrados de SDS-PAGE en gel en un PVDF o nitrocelulosa membrana. Proteínas transferidas se analizaron para las modificaciones post-traduccionales utilizando sondas que pueden detectar los lípidos , hidratos de carbono , fosforilación o cualquier otra modificación de proteínas. Este Blot se debe usar para referirse a métodos que detectan sus objetivos a través de la interacción específica de la PTM y la sonda, distinguiéndolos de un estándar de transferencia Far-Western . En principio, este secante es similar a lectina secante (es decir, la detección de epítopes de carbohidratos en las proteínas o lípidos).
ALESHA LOW 2012-1318
ResponderEliminarWESTERN BLOT PARA PROTEINAS
El Western blot, o inmunoblot, es una técnica analítica usada para detectar proteínas específicas en una muestra determinada (una mezcla compleja de proteínas, como un extracto tisular). Mediante una electroforesis en gel se separan las proteínas atendiendo al criterio que se desee: peso molecular, estructura, hidrofobicidad, etc. Hay casi tantas posibilidades como tipos de electroforesis existen. Luego son transferidas a una membrana adsorbente (típicamente de nitrocelulosa o de PVDF) para poder buscar la proteína de interés con anticuerpos específicos contra ella.Finalmente, se detecta la unión antígeno-anticuerpo por actividad enzimática, fluorescencia entre otros métodos. De esta forma se puede estudiar la presencia de la proteína en el extracto y analizar su cantidad relativa respecto a otras proteínas.
Esta técnica es hoy en día imprescindible en varios campos de la biología, como la biología molecular, la bioquímica, la biotecnología o la inmunología. Existe toda una industria especializada en la venta de anticuerpos (monoclonales y policlonales) contra decenas de miles de proteínas diferentes.
El Western blot fue desarrollado en el laboratorio de George Stark, en la Universidad de Stanford. El nombre (Western, occidental en inglés) le fue dado por W. Neal Burnette, y consiste en un juego de palabras con una técnica análoga pero que usa DNA, el Southern (sureño) blot, que en este caso debe su nombre a su descubridor, Edwin Southern. Otras técnicas que fueron nombradas siguiendo este criterio son el Northern blot (en el que se separa e identifica RNA), el Eastern blot, el Southwestern blot.
ALESHA LOW 2012-1318
ResponderEliminarSOUTHWESTERN BLOT
El Southwestern blot es una técnica de laboratorio que permite la identificación y caracterización de proteínas por su capacidad de unión a fragmentos específicos de ADN. Para ello, se transfiere el resultado de una electroforesis a una membrana y se añaden oligonucleótidos para observar la unión a las proteínas. Fue descrito por primera vez en 1981 en el laboratorio de Brian Bowen.
Su nombre se debe a la convergencia entre el Southern blot, que emplea sondas de ADN para detectar, en su caso, ADN fijado en una membrana, y el Western blot, en el cual se detectan proteínas fijadas a una membrana, aunque en este caso se realice con anticuerpos.
ALESHA LOW 2012-1318
ResponderEliminarEXTRACCION DE DNA MITOCONDRIAL
También llamado cromosoma mitocondrial, es una molécula circular de DNA (*) de un tamaño de 16569 pares de bases (bp) (8000 veces menor que el cromosoma medio). Este tamaño de 16569 bp corresponde al primer ADN secuenciado (secuencia Cambridge) aunque existen otras variantes con un número de pares de bases que oscila entre 16559 y 16570. En cada mitocondria existen varias copias de este ADN, de modo que el número de cromosomas mitocondriales en cada célula puede ser de varios miles. Cuatro o cinco cromosomas mitocondriales se agrupan formando los llamados nucleoides
El ADN mitocondrial es muy parecido al del los cromosomas bacterianos, estando formado por dos cadenas complementarias, cada una con 16569 pares de bases, pero con un peso molecular diferente: la cadena pesada H (peso molecular, 5.168.726 daltons) contiene muchas más G que la cadena ligera L (peso molecular, 5.060.609 daltons). La mayor parte de la cadena H constituye el molde para la transcripción de la mayor parte de los genes, mientras que la cadena L es la cadena codificadora (*)
El genoma mitocondrial contiene un total de 37 genes de los cuales 13 genes que codifican para ARNs mensajeros, y por lo tanto para 13 proteínas, 22 genes que codifican para 22 tARNs (ARNs de transferencia, que se representan simbólicamente como hojas de trebol) (*) y 2 genes que codifican para dos rRNAs mitocondriales (RNAs ribosómicos). En la práctica, cuando se secuencia el ADN miocrondrial se unna persona en particular, se observan un cierto número de variaciones que son simplemente polimorfismos no tienen ninguna consecuencia clínica. De hecho, una región del ADN-mitocondrial llamada región de control, es tan polimórfica que se utiliza en medicina forense para identificar al sujeto.
Además de las proteínas que la mitocondria puede sintetizar por si misma, necesita importar algunas otras sintetizadas en el núcleo. De igual forma, los lípidos que forman las membranas externa e interna de la mitocondria son importadas.
Las mutaciones de algunos de los genes mitocondriales ocasionan enfermedades en el hombre. Se conocen las siguientes mutaciones:
MELAS: (miopatía mitocondrial con encefalopatía, acidosis láctica y episodios similares al ictus). Se debe a una disfunción el complejo I de la cadena respiratoria mitocondrial debida a un cambio de bases en el par 3243 de la cadena pesada
MERRF: (epilepsia mioclónica, fibras rojas deshilachadas): se debe sobre todo a una mutación del gen que codifica el t-ARN de la lisina por un cambio de bases en la posición 8344 de la cadena pesada. Este cambio produce una disfunción del complejo V de la cadena respiratoria
NARP (neuropatía, ataxia, retinitis pigmentaria): se debe a una mutación del gen que codifica el complejo V de la cadena respiratoria (ATP-asa 6)
LHON (neuropatía hereditaria de Leber): se debe a multiples mutaciones en los genes que codifican el complejo I (NADH-deshidrogenasa)
Adicionalmente se han encontrado estas y otras mutaciones de los genes mitocondriales en muchos otros desórdenes (p. ejemplo, sordera, síndrome de Ham, etc).
ALESHA LOW 2012-1318
ResponderEliminarELECTROFORESIS DE PROTEINAS.
La electroforesis se utiliza para identificar la presencia de proteínas anómalas, para identificar si falta alguna de las proteínas normales y para determinar si diferentes grupos de proteínas en sangre están aumentados o disminuidos.
Alteraciones del patrón obtenido característicamente permiten orientar el diagnóstico hacia una enfermedad u otra. La presencia de una anomalía en un patrón electroforético raramente es diagnóstica por sí misma, aunque sí proporciona una pista. El estudio en estos casos se ampliará con la finalidad de identificar la naturaleza de la enfermedad subyacente.Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina).
Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (-) o cátodo (+) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración.
Isoelectroenfoque: en lugar de separar las proteínas en función de su carga a un pH dado, se separan en función de su punto isoeléctrico (pI): el pI es el pH en el que la carga neta de la proteína es nula, y depende de la composición aminoacídica de la proteína. Se crea un gradiente de pH mediante anfolitos (estabilizan el pH a lo largo del gel). Cada proteína migrará hasta alcanzar su pI, punto en el cual precipitará al acumularse (de ahí el nombre, isoelectroenfoque).
También se suelen utilizar acoplados a zimogramas si deseamos determinar el pI de una enzima o en electroforesis bidimensional como primera dimension.
Separación por tamaño: permite separar proteínas y ácidos nucleicos. En el caso de las proteínas, deben ser tratadas con SDS (sodio dodecil sulfato) para que su carga sea negativa y todas migren hacia el ánodo (no es necesario hacer eso con los ácidos nucleicos, ya que tienen carga negativa); la separación se hace en medios de soporte en el que se ha creado un tamiz molecular (matriz), que hace que las proteínas más pequeñas corran más que las más grandes.
Tema 2 :Reacción de cadena polimerasa (PCR)
ResponderEliminarLa reacción en cadena de la polimerasa, ya más conocida como PCR, es una técnica que permite replicar entre cientos de miles y millones de veces, en el transcurrir de pocas horas e in vitro, pequeñas cantidades de ADN. Uno de los aportes fundamentales de esta metodología a la investigación básica en biología molecular consiste, precisamente, en la rapidez y eficiencia mediante las cuales se realiza una tarea que antes requería largas y tediosas jornadas. El producto quese obtiene al finalizar la reacción es una gran cantidad de un fragmento génico con alto grado de pureza- favorece la tarea de los investigadores empeñados en ampliar nuestros conocimientos sobre la estructura y función de los genes. El método se basa, en su forma más simple en la realización de tres reacciones sucesivas llevadas a cabo a distintas temperaturas. Estas reacciones se repiten cíclicamente entre veinte y cuarenta veces (véase la figura 1). La muestra se calienta, en el primer paso, hasta lograr la separación de las dos cadenas que constituyen el ADN, hecho que se conoce como "desnaturalización". En el segundo paso, la temperatura se reduce para permitir el "apareamiento" de cada una de dos cadenas cortas de nucleótidos (oligonucleóridos) con cada una de las hebras separadas del ADN molde. Se trata de segmentos de ADN de cadena simple, sintetizados en el laboratorio y diseñados de manera tal que permiten definir los límites del tramo de ADN que se desea replicar. Obviamente, para que se pueda producir el apareamiento, cada uno de estos oligonucleótidos, a los que se denomina "iniciadores" o primers, debe ser complementario del tramo al que tienen que unirse en las cadenas separadas del ADN molde. En tercer lugar, una enzima ADN polimerasa extiende los primers, en el espacio comprendido entre ambos, sintetizando las secuencias complementarias de las hebras del ADN molde. Para ello, la ADN polimerasa usa desoxidonucleósidos trifosfato agregados a la mezcla de reacción. La temperatura a la que se realiza el tercer paso está condicionada por aquélla a la cual "trabaja" la enzima ADN polimerasa. Al cabo del primer ciclo de tres reacciones (desnaturalización, apareamiento, extensión) el tramo de ADN elegido se ha duplicado y el doble de su cantidad original se encuentra disponible para ser nuevamente replicado en un segundo ciclo. El resultado de la aplicación de numerosos ciclos "en cadena" da lugar a la amplificación geométrica del segmento de ADN delimitado por los primers.Para replicar el ADN, la técnica PCR hizo uso, en un principio, de la ADN polimerasa de la bacteria Escherichia coli. Pero esta enzima resulta desactivada debido a la alta temperatura requerida para desnaturalizarla doble cadena del ADN, por lo cual debía agregarse enzima fresca al comenzar el tercer paso de cada ciclo. Este inconveniente fue solucionado de manera ingeniosa cuando se la reemplazó por su equivalente de la bacteria "termófila" Thermus aquaticus. En efecto, la ADN polimerasa de este microrganismo, denominada Taq polimerasa, actúa eficientemente entre los 75° C y los 80° C y resiste más de dos horas a 93° C. De esta manera es posible mezclar todos los componentes de la PCR al comenzar el primer ciclo y la reacción en cadena puede llevarse a cabo mediante equipos automatizados que realizarán los ciclos térmicos programados.
2012-1309
ResponderEliminarTema 3: Enzimas de restrincion (BRENDA)
Las enzimas de restricción son proteínas cuya función es cortar las hebras de ADN. Se podría decir que son “tijeras moleculares” que cortan ADN. Lo hacen en forma específica. Esto significa que cada enzima reconoce un sitio particular del ADN, es decir que reconoce una secuencia particular de nucleótidos. Esa secuencia específica para cada enzima se denomina “sitio de restricción”. Una vez que la enzima reconoce estos sitios, se posiciona sobre la molécula de ADN y corta dentro o en torno de esa secuencia. Las enzimas de restricción, conocidas también como endonucleasas, sólo cortan el ADN si reconocen en su interior una secuencia específica de nucleótidos. Estas enzimas fueron descubiertas en microorganismos. De hecho, se encuentran sólo en organismos procariotas (bacterias). Por esto, se les dio una nomenclatura asociada al organismo de donde provienen. Por ejemplo, las enzimas de restricción que se descubrieron en la bacteria Escherichia coli se denominan Eco. Existen diferentes tipos de enzimas Eco que se diferencian en la secuencia que reconocen y cortan. Para diferenciarlas se les agregan letras y números romanos, por ejemplo: Eco RI (Eco “erre” “uno”). Así, el sitio de restricción para EcoRI es la secuencia GAATTC, como muestra la ilustración anterior. Una vez que la enzima encontró ese sitio en el ADN, se acerca a la hebra de ADN y realiza el corte entre la G y la A. Al investigar otras especies de bacterias se descubrieron cientos de enzimas de restricción distintas y cada una reconoce una región específica. Estas enzimas fueron descubiertas en la década del ’70 y hasta la fecha existen más de 250 enzimas de restricción. Se cree que la función natural de estas enzimas en las bacterias es protegerlas contra ADN de virus que podrían ingresar en sus células. De esta manera, la bacteria utiliza estas “tijeras moleculares” para cortar en pedacitos el ADN viral que la infecta. El ADN propio de la bacteria no se corta, pues lo tiene “protegido” contra sus propias enzimas de restricción.
2012-1309
ResponderEliminarTema 4: ELISA directo (Sandwish)
La técnica de ELISA (Enzyme Linked InmunoSorbent Assay) es un procedimiento de ensayo inmuno enzimático. Se basa en la detección antígeno(Ag) o anticuerpo (Ac), La técnica de ELISA es un ensayo inmunoenzimático ampliamente empleado en el área médica para la cuantificación de moléculas especialmente de aquellas que experimentan cambios en diferentes estados como pueden ser infecciones por bacterias, virus, hongos o parásitos o fases activas de enfermedades autoinmunes. Como ejemplo se pueden medir por ELISA hormonas, autoanticuerpos, inmunoglobulinas contra antígenos de patógenos, toxinas, antígenos. Las técnicas de ELISA son también muy usadas en los ensayos de nuevos medicamentos para comprobar sus efectos cuantificando las moléculas de interés en cada caso. Existen numerosas variantes de este tipo de ensayo. Entre ellos se encuentran los métodos directo e indirecto. El método directo permite la detección del antígeno con un anticuerpo específico conjugado con una enzima como sistema de marcaje. Técnica: Las placas ELISA se preparan recubriendo los pocillos con las soluciones en las que se sospecha se encuentra el antígeno. Se incuban con anticuerpos marcados. Indican la presencia de antígeno en la solución analizada. Es necesario incluir controles negativos que serán muestras del mismo tipo de las analizadas (sangre, orina) pero en las que se tenga la certeza de la ausencia del antígeno buscado. Asimismo se incluyen controles positivos (soluciones donde se encuentra el antígeno buscado).
Elisa sandwish Se trata de un ensayo muy empleado en el que se recubre el pocillo con un primer anticuerpo anti-antígeno. Después de lavar el exceso de anticuerpo se aplica la muestra problema en la que se encuentra el antígeno, que será retenido en el pocillo al ser reconocido por el primer anticuerpo. Después de un segundo lavado que elimina el material no retenido se aplica una solución con un segundo anticuerpo anti-antígeno marcado. Así pues cada molécula de antígeno estará unida a un anticuerpo en la base que lo retiene y un segundo anticuerpo, al menos, que lo marca. Este ensayo tiene una gran especificidad y sensibilidad debido a la amplificación de señal que permite el segundo anticuerpo.
2012-1309
ResponderEliminarTema 5:ELISA Indirecto
En el método indirecto el antígeno reacciona con el anticuerpo específico. El complejo antígeno-anticuerpo es entonces detectado por un segundo anticuerpo que reconoce dominios constantes de anticuerpos. Este anticuerpo, que suele ser específico de especie, es el que está marcado enzimáticamente. Esto permite que un mismo anticuerpo marcado sea capaz de detectar diferentes antígenos. En ambos métodos, la reacción enzimática puede ser detectada espectrofotográficamente con un lector ELISA.
Es el ensayo más popular, como lo es la inmunofluorescencia indirecta, pues un mismo secundario marcado y un mismo sistema enzimático permite cuantificar una gran variedad de antígenos, por eso es un método más polivalente y barato, aunque se pierda algo de precisión por tener un eslabón más con respecto al método directo. La dilución de la solución que contiene el anticuerpo primario (por ejemplo: suero sanguíneo) es un factor muy importante a tener en cuenta para evitar la aparición de falsos negativos, ya que si la muestra está muy diluida nosaldrá positiva si la titulación de anticuerpos es muy baja. Es decir, aunque los anticuerpos están presentes, la prueba no da positivo porque la concentración de anticuerpos específicos contra el antígeno que está pegado en el fondo del pocillo no es suficiente como para dar una señal detectable.El ELISA indirecto es el método de elección para detectar la presencia de anticuerpos séricos contra el virus de la inmunodeficiencia humana (VIH), agente causante del síndrome de inmunodeficiencia adquirida (SIDA). Según esta técnica, proteínas recombinantes de la envoltura y el núcleo del VIH se absorben como antígenos en fase sólida a los pocillos. Las personas afectadas de VIH producen anticuerpos séricos contra epítopos en estas proteínas víricas. En general, el ELISA indirecto permite detectar anticuepos séricos contra VIH desde las primeras seis semanas de una infección.
2012-1309
ResponderEliminarTema 6:Tecnicas Blotting
El término “blotting” hace referencia a la transferencia de macromoléculas biológicas desde un gel hasta una membrana y su posterior detección en la superficie de la misma. Este proceso es necesario ya que todas estas macromoléculas están embebidas dentro de la matriz que forma el gel, lo que imposibilita realizar su detección, mientras que en la membrana, se encuentran accesibles al estar adheridas sobre la superficie de la misma. La técnica del Western Blot (también llamado “inmunoblotting” debido a que se utilizan anticuerpos para detectar el antígeno o los antígenos específicos de interés) fue descrita por primera vez por Towbin et al. en 1979 y en la actualidad es una técnica de rutina en todos los laboratorios que realizan análisis de proteínas. La especificidad de la unión antígeno – anticuerpo permite la detección de una única proteína dentro de una mezcla compleja de otras proteínas. En la actualidad se utiliza como criterio de identificación positivo de una proteína especifica en una mezcla compleja y para obtener datos cualitativos y semicuantitativos sobre la misma.
2012-1309
ResponderEliminarTema 7:Electroforesis de proteínas
Es un método de separación de proteínas mediante la aplicación de un campo eléctrico. Existen diferentes tipos en función del tipo de separación empleado: electroforesis de zona (separación en función de la carga), isoeléctrico enfoque y separación por tamaño en tamiz molecular (también aplicable a ácidos nucleicos). Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina).Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (-) o cátodo (+) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración.
2012-1309
ResponderEliminarTema 8:El ADN mitocondrial
Es un fragmento de ácido nucleico encontrado dentro de las mitocondrias. Las mitocondrias son pequeños organelos o estructuras celulares que se encuentran dentro de las células eucarióticas (generalmente las células animales). Estos organelos se usan básicamente para la producción y generación de energía para que la célula lleve a cabo sus funciones. Las plantas tienen cloroplastos. Las células animales tienen mitocondrias. Y cada una de estas mitocondrias tiene su propio genoma. Su propia molécula circular de ADN. Y eso es el ADN mitocondrial, porque en realidad es diferente del ADN que se encuentra en el núcleo de las células. No se encuentra dentro del núcleo. El número de genes en el ADN mitocondrial es de 37, frente a los 20.000 - 25.000 genes del ADN cromosómico nuclear humanos.Codifica dos ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa. El ADN mitocondrial está en replicación constante, independientemente del ciclo y del tipo celular. Se piensa que tiene lugar de forma asíncrona, es decir, que tiene lugar en las dos cadenas en tiempos diferentes y con dos orígenes distintos hacia direcciones contrarias. El comienzo tendría lugar en el origen de la cadena pesada, situado en el bucle D, y replicaría ésta tomando como molde la cadena ligera. Cuando se alcanza el segundo origen, situado a dos tercios de distancia del primero, comienza la segunda ronda de replicación en sentido opuesto. Se ha propuesto un nuevo sistema de replicación que coexistiría con el primero. Sería bidireccional y comportaría una coordinación entre hebras directas y retrasadas.
2012-1309
ResponderEliminarTema 9: Anticuerpos monoclonales
Son anticuerpos homogéneos producido por una célula híbrida producto de la fusión de un clon de linfocitos B descendiente de una sola y únicacélula madre y una célula plasmática tumoral. Los anticuerpos monoclonales mAB, de la frase en inglés con idéntico significado que en español: monoclonal antibody, son anticuerpos idénticos porque son producidos por un solo tipo de célula del sistema inmune, es decir, todos los clones proceden de una sola célula madre. Es posible producir anticuerpos monoclonales que se unan específicamente con cualquier molécula con carácter antigénico. Este fenómeno es de gran utilidad en bioquímica, biología molecular y medicina. Si una sustancia extraña (antígeno) se inyecta en el cuerpo de un ratón o un humano, alguna de las células B de su sistema inmune se transformarán en células plasmáticasy empezarán a producir anticuerpos que se unirán a ese antígeno. Cada célula B produce un solo tipo de anticuerpo, pero diferentes linfocitos B producirán anticuerpos estructuralmente diferentes que se unen a distintas partes del antígeno. Esta mezcla fisiológica natural de anticuerpos es conocida como Antisuero policlonal.Para producir anticuerpos monoclonales, primero se extraen células B del bazo de un animal que ha sido expuesto al antígeno. Estas células B son fusionadas en presencia de PEG (polietilenglicol) con células tumorales de mieloma múltiple (un tipo de cáncer) que pueden crecer indefinidamente en cultivo celular. Esta fusión hace a las membranas celulares más permeables. Estas células fusionadas híbridas, llamadas hibridomas pueden multiplicarse rápida e indefinidamente, puesto que son células tumorales después de todo y pueden producir gran cantidad de anticuerpos. Los hibridomas son suficientemente diluidos y cultivados para obtener un número diferente de determinadas colonias, las cuales producen sólo un tipo de anticuerpo. Los anticuerpos de diferentes colonias son analizados para conocer su capacidad de unirse a un antígeno determinado, por ejemplo con un tipo de test llamado ELISA, y para seleccionarse y aislarse de la manera más efectiva.